
SAM文件与Samtools
SAM文件
SAM (sequence alignment format) 是一种专门用于储存序列比对信息的文件格式,BAM文件是SAM的二进制文件。
当测序生成的fastq文件比对到参考基因组后就会生成SAM文件或者BAM文件。
SAM文件包括头部注释和比对结果两部分,头部注释为''可选部分''。
头部注释位于比对结果之前,以“@”开头。比对结果有11列是固定的,其他多列可选。
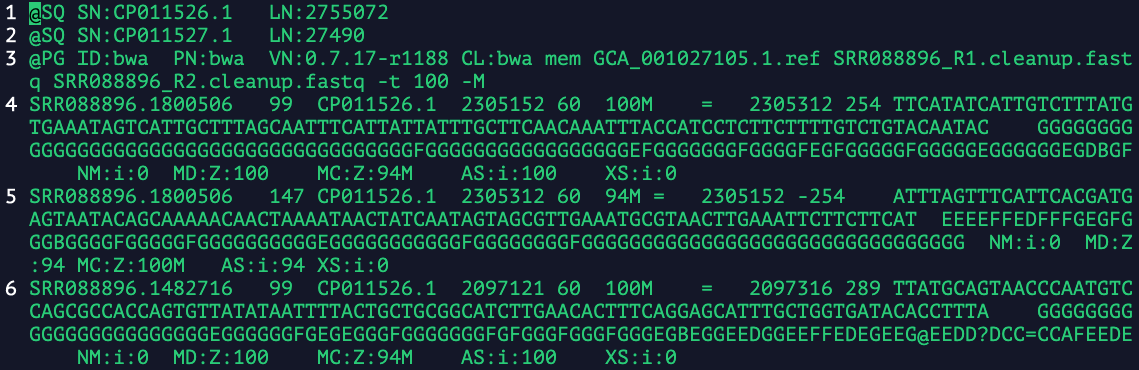
头部注释部分
在上图的示例中,前三行为头部注释。
- @SQ:参考序列目录;SN: 参考序列名字;LN: 参考序列长度。
- @PG:使用的比对程序名,这个例子是bwa;VN: bwa的版本;CL: 执行程序的命令
有时还会有 (推荐的规范):
@HD VN:1.0 SO:unsorted
VN是格式版本;SO表示比对排序的类型,有unkown (default),unsorted,queryname和coordinate几种。
比对结果部分
从第四行开始为比对结果,自左向右,前11列为固定字段,分别为:
- 第1列 Query Name:查询短序列的名称,即read的编号。
- 第2列 FLAG:如果不是以下数字中的一个,则是某几项数据的和。
1:表示read有多个测序数据,一般理解为有双端测序数据,另一条没有过滤掉;2:表示read的多个片段都有比对结果,双端的read都能比对上;4:表示该read没有比对上;8:表示下一条read没有比对上;16:表示该read的反向比对上了;32:表示该read的下一条的反向没有比对上;64:表示样本中第一条片段;128:表示样本中最后一条片段;256:表示第二次比对;512:表示比对的质量不合格;1204:表示read是PCR或光学副本产生的;2048:表示辅助比对结果;
- 第3列 Reference Name:参考序列的名称,或者比对到参考序列上的染色体号。比对不上为
*。 - 第4列 Position:比对上的位置,从1开始计数 (顺着链的方向从1数起), 没有比对上为
0; - 第5列 Mapping Quality:比对的质量分数,越高表示比对的越准确。
- 第6列 CIGAR:表示比对的结果。详见序列匹配中的CIGAR
- 第7列 RNEXT:表示下一个片段比对上的参考序列的编号,比对不上用
*,该片段和下一个片段比对上同一个参考片段,用=; - 第8列 PNEXT:表示下一个片段比对上的位置,如果不可用,此处为0;
- 第9列 TLEN:表示Template的长度。如果第八列大于第四列,则为正数,否则负数。
- 第10列 SEQ:表示序列片段的序列信息,(注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度),表示read的碱基序列,如果是比对到互补链上则是反转互补序列。
- 第11列 QUAL:表示read的质量,用ASCII编码表示。
可选字段以TAG:TYPE:VALUE的形式提供额外的信息。
Samtools
samtools是一个用于操作SAM和BAM文件的工具合集。BAM文件优点:BAM文件为二进制文件,占用的磁盘空间比SAM文本文件小;且基于BAM二进制文件的运算速度快。
samtools view
功能:将SAM文件转换成BAM文件,以便于对BAM文件进行各种操作。
用法: samtools view [options]
默认情况下不加region,则是输出所有的regions。
- -b 默认下输出是SAM格式文件,该参数设置输出BAM格式
- -h 设定输出SAM文件时带
头部注释,默认时不输出;-H 仅输出头部注释,无比对结果 - -S 默认下输入是BAM文件,若是输入是SAM文件,则最好加该参数,否则有时候会报错
- -u 输出非压缩的BAM文件,需要有-b参数,能节约时间,但是需要更多磁盘空间
- -1 输出快速压缩的BAM文件,需要有-b参数
- -c 不输出
比对结果,而仅计数 - -F INT 过滤FLAG等于该参数的比对结果
例子:
# samtools view -b -S abc.sam -o abc.bam
将SAM文件转换成BAM文件。
# samtools view -b -F 4 abc.bam > abc.F.bam
提取比对到参考序列上的比对结果。
# samtools view -b -F 12 abc.bam > abc.F12.bam
提取paired reads中两条reads都比对到参考序列上的比对结果,只需把两个4+8的值12作为过滤参数。
# samtools view -bf 4 abc.bam > abc.f.bam
提取没有比对到参考序列上的比对结果。
# samtools view abc.bam scaffold1 > scaffold1.sam
提取BAM文件中比对到scaffold1 (作为region名称) 上的比对结果,并保存到SAM文件格式。
# samtools view abc.bam scaffold1:30000-100000 > scaffold1_30k-100k.sam
提取scaffold1上能比对到30k到100k区域的比对结果。
samtools sort
功能:对BAM文件进行排序。
用法: samtools sort [-n][-m ] <in.bam> <out.prefix>
- -m 参数默认下是 500,000,000,不支持K,M,G等缩写,即默认值不能设为500M。当处理大数据时,如果内存够用,则设置较大值,可节约时间。
- -n 设定排序方式按short reads的ID排序。默认下是按序列在fasta文件中的顺序(即header)和序列从左往右的位点排序。
例子:
# samtools sort abc.bam abc.sort
对abc.bam进行排序,输出文件为abc.sort.bam。
samtools index
功能:对BAM文件建立索引,并生成后缀为.bai的文件,用于快速的随机处理。很多情况下需要有bai文件的存在,特别是显示序列比对情况下,比如samtool的tview命令。
用法:samtools index <in.bam> [out.index]
例子:
# samtools index abc.sort.bam abc.sort.bam.bai
生成abc.sort.bam的索引文件abc.sort.bam.bai,输出文件名可省略。
samtools faidx
功能:对fasta文件建立索引,并生成后缀为.fai的文件。该命令也能依据索引文件快速提取fasta文件中的某一条子序列。
用法:samtools faidx <in.bam> [ [...]]
例子:
# samtools faidx genome.fasta
# samtools faidx genome.fasta scaffold_1 > scaffold_1.fasta
从genome.fasta中提取名为scaffold_1的字序列。
samtolls tview
功能:直观的显示出reads比对基因组的情况。
用法:samtools tview <aln.bam> [ref.fasta]
如给出参考基因组,会在第一排显示参考基因组的序列,否则,第一排全用N表示。显示界面下操作指令的使用说明,可按?键来查看。
samtools mpileup
功能:用于生成bcf文件,再使用bcftools进行SNP和Indel的分析。bcftools是samtool中附带的软件。
用法:samtools mpileup [-EBug][-C capQcoef] [-r reg][-f in.fa] [-l list][-M capMapQ] [-Q minBaseQ][-q minMapQ] in.bam [in2.bam]
- -f 指定有索引文件的fasta参考序列
- -g 输出bcf格式
- -D Output per-sample read depth (require -g/-u)
- -S Output per-sample Phred-scaled strand bias P-value (require -g/-u)
例子:
# samtools mpileup -f genome.fasta abc.bam > abc.txt
mpileup不使用-u或-g参数时,则不生成二进制的bcf文件,而生成一个文本文件(输出到标准输出)。
结果包含6列:参考序列名;位置;参考碱基;比对上的reads数;比对情况;比对上的碱基的质量。
# samtools mpileup -g -S -D -f genome.fasta abc.bam > abc.bcf