
序列匹配中的CIGAR
CIGAR (Compact Idiosyncratic Gapped Alignment Report),是SAM/BAM格式表示序列对齐的方式。了解CIGAR串将帮助我们了解查询 (Query) 序列如何与参考基因组 (Reference) 对齐。
构成CIGAR的编码方式有:
| SAM | BAM | Description |
|---|---|---|
| M | 0 | 比对匹配 (可以是序列匹配或不匹配) |
| I | 1 | 查询序列中发生碱基插入,即在参考序列中插入空缺 |
| D | 2 | 查询序列中发生碱基缺失,即在参考序列中插入碱基 |
| N | 3 | 在参考序列中的跳过,与D类似,但长度较长,或称为跳跃区 |
| S | 4 | 软剪切 (被剪切的序列信息仍保存于SAM/BAM文件) |
| H | 5 | 硬剪切 (被剪切的序列信息不再保存于SAM/BAM文件中) |
| P | 6 | padding (silent deletion from padded reference) |
| = | 7 | 序列匹配 |
| X | 8 | 序列不匹配 |
Example 1.

CIGAR: 6M
解释:从reference的第3位开始,query的6个碱基都与reference匹配。
Example 2.

CIGAR: 3M2I3M
解释:从reference的第3位开始,query有3个碱基与reference匹配,然后有2个碱基插入,再有3个碱基匹配。
Example 3.

CIGAR: 2M1D3M
解释:从reference的第3位开始,query有2个碱基与reference匹配,然后有1个碱基缺失,再有3个碱基匹配。
Example 4.

CIGAR: 3M7N4M
解释:从reference的第4位开始,query有3个碱基与reference匹配,然后有7个碱基缺失,再有4个碱基匹配。
Example 5.
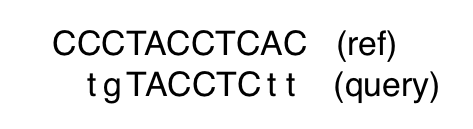
CIGAR: 2S6M2S
解释:查询序列两侧分别有2个碱基的被切掉,但在SAM/BAM文件中仍保留被剪切掉的碱基序列(SAM文件第10列SEQ)。而应剪切H,则在SAM文件第10列SEQ中不保留。
N 主要发生在查询序列夸内含子匹配到 (双侧) 外显子的情形。
M 编码的匹配包括匹配match与错配mismatch。
H 主要用于标记嵌合比对 (同一查询序列匹配到参考序列的不同位置,非最优比对通常定义为嵌合比对),其中最优比对 (top hit) 会用S,而其他非最优比对会用H。
X & = 在主流序列匹配软件已不常用。
在不支持多序列比对的软件中,P可忽略。
cigar (Python包) 可用于CIGAR的解析。
使用CIGAR的常用软件有:USEARCH,bowtie2, bwa, minimap2
网络上很多关于CIGAR的解释多为“人云亦云”,需谨慎阅读。