
宏基因组数据分析——metaWRAP
1. metaWRAP简介
MetaWRAP旨在成为一个易于使用的宏基因组数据分析软件包,从头到尾完成宏基因组分析的核心任务:序列质量控制、组装、可视化、分类分析、提取基因组草图(又称分箱binning)和功能注释。此外,metaWRAP将bin提取和分析提升到了一个新的层次(参见下面的模块概述)。虽然没有简单的最佳方法来处理宏基因组数据,但在深入研究分析参数之前,metaWRAP是一种快速而简单的方法。MetaWRAP可应用于多种环境,包括肠道、水和土壤微生物组(详情请参阅MetaWRAP手册)
宏基因组 ( Metagenome)(也称微生物环境基因组Microbial Environmental Genome, 或元基因组)是由 Handelsman 等 1998 年提出的新名词, 其定义为“the genomes of the total microbiota found in nature” , 即环境中全部微小生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因。
2. metaWRAP的分析模块
通过metawrap -h命令可显示metawrap命令的帮助信息,其中重点在于其能完成的功能模块,包括:
- read_qc # 数据质控模块,包括去除宿主/污染序列。
- assembly # 拼接模块,由短序列reads拼接成重叠群contigs。
- binning # 重叠群分箱模块,将重叠群contigs根据四联密码子相似性分为不同的组,表示一个分类单元的基因组。
- bin_refinement # 分箱提炼模块,根据metaBAT2,MaxBin2,CONCOCT三软件的结果,整合最优分箱。
- reassemble_bins # 分箱重组装模块,根据分箱结果,将短序列reads重新组装,以期优化组装结果。
- quant_bins # 分箱丰度分析模块。
- kraken # 短序列reads/重叠群contigs分类学注释模块。
- blobology # 数据可视化模块。
- annotate_bins # 分箱基因功能注释模块。
- classfy_bins # 分箱分类学注释模块。
metaWRAP 模块的逻辑关系如下图所示:
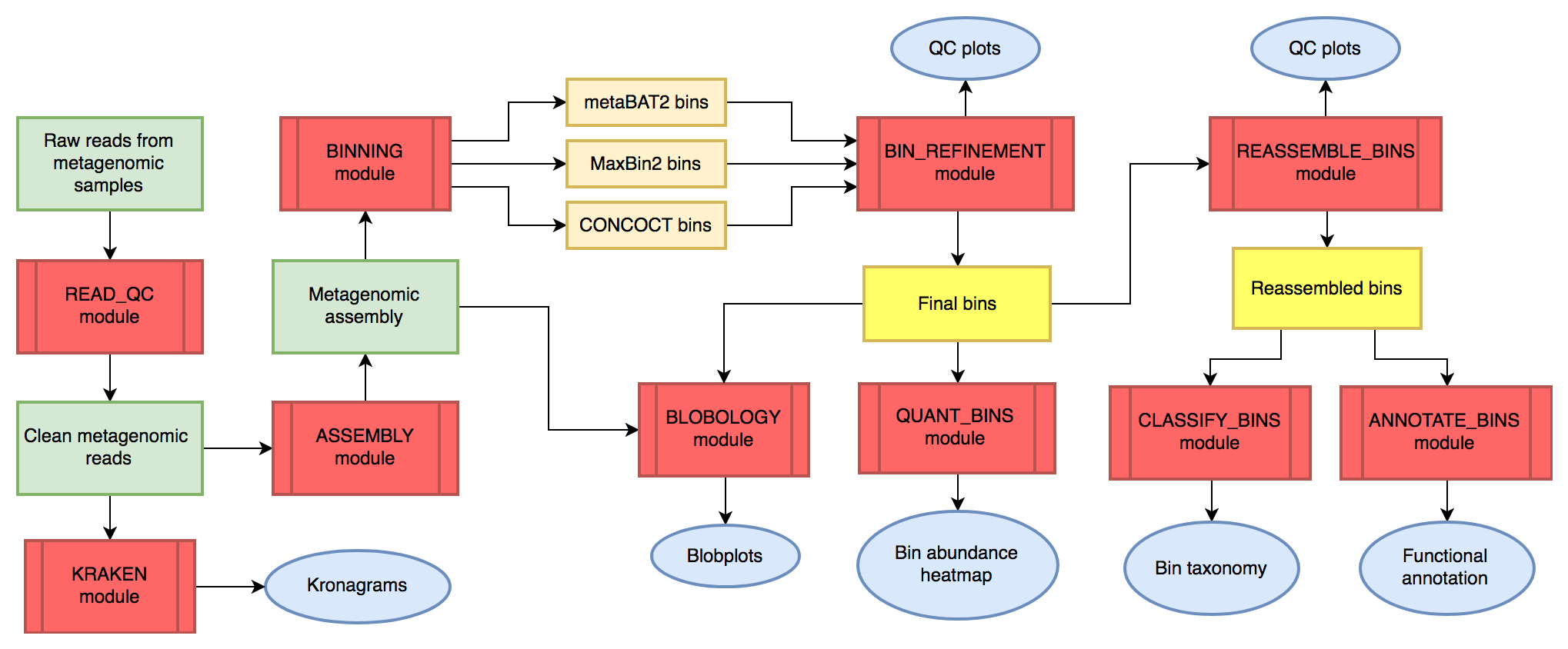
其中,绿色板块和黄色板块代表数据,红色板块代表metaWRAP的分析模块,灰蓝板块代表数据可视化结果。
数据流上按照以下逻辑关系组织:
**原始reads数据->质控后的clean reads数据->拼接后的重叠群contigs数据->分箱后的bins数据->分类学、基因功能等综合分析结果 **
3. metaWRAP的使用
3.0 数据准备
宏基因组数据按照来源可分为自产数据,公共数据和模拟数据。
3.0.1. 自产数据
根据实验设计,采集样品,提取环境总DNA,送交测序服务公司测序。
3.0.2. 公共数据
方法1:利用一般下载工具(需首先获得数据资源的地址)
(sradownload) [user@server ~]# wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR5031889/ERR5031889_1.fastq.gz
(sradownload) [user@server ~]# wget ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR5031889/ERR5031889_2.fastq.gz
(sradownload) [user@server ~]# gzip -d *.gz
方法2:利用NCBI sratools下载
(sradownload) [user@server ~]# prefetch ERR5031889 # 根据给定的run accession下载
(sradownload) [user@server ~]# fastq_dump --split-files ERR5031889.sra # 将sra格式转为fastq
方法3:利用enaBrowserTools下载
(enatools) [user@server ~]# enaDataGet -a -f 'fastq' -as /path/to//key_files/aspera_settings.ini ERR3021563 # 参数-as : 指定Aspera密匙
3.0.3. 模拟数据
InSilicoSeq是一个产生Illumina短序列数据的模拟器。主要用于模拟宏基因组样本,也可用于从单个基因组产生测序数据。
(insilicoseq) [user@server ~]# iss generate --ncbi bacteria -U 30 --model miseq --output miseq_ncbi --n_reads 10m --cpus 24
# 从NCBI细菌数据库中选取30个物种(随机选取,也可以指定物种以及丰度)来生成10m reads的双末端MiSeq数据,生成的两个文件各含有500万个reads。
模拟数据在方法开发,以及数据分析可重复性评价上有重要作用。除InSilicoSeq外,还有CAMISIM可供选择。
以下分析流程基于InSilicoSeq的模拟数据
3.1 数据质控 (read_qc模块)
高通量测序的建库过程中,由于各种物理化学原因或者污染,以及测序仪本身的问题,都会导致问题序列的产生。所以高通量测序数据的质控主要包括去除接头adapter,过滤低质量reads,以及涉及宿主时,去除宿主来源的reads。
metawrap read_qc借助Trim-galore(默认参数),根据接头序列和PHRED评分 (阈值为20) 对原始数据raw data进行处理,以保留高质量的clean reads。然后clean reads会被BMTagger比对到人基因组 (需要配置相应数据库),并去除 (配对reads中如仅有一端比对到宿主序列上,也会被去除)。最后,FASTQC用于生成raw reads和clean reads的质量报告,以评估reads质量改进。
命令:
(metawrap-env) [user@server ~]# metawrap read_qc -1 raw_data/10m_1.fastq -2 raw_data/10m_2.fastq -t 96 -o read_qc --skip-bmtagger
参数说明:
- -1 正向双末端测序fastq文件
- -2 反向双末端测序fastq文件
- -t 计算线程数
- -o 结果输出目录
- --skip-trimming # 跳过trim Galore的剪切
- --skip-bmtagger # 跳过去宿主操作
- --skip-pre-qc-report # 跳过对raw reads的FastQC评价
- --skip-post-qc-report # 跳过clean reads的FastQC评价
非人类宿主源片段的去除,可参考用Bowtie2去宿主序列
3.2 拼接 (assembly模块)
宏基因组研究主要有两种策略,一种是当有参考基因组,直接将reads比对到参考基因组上进行研究。其次是当没有参考基因组信息时,需要通过序列组装,基因预测,后续物种注释以及功能注释。而多数情况下,宏基因组数据分析为无参分析。
metawrap assembly借助metaSPAdes和MegaHit完成对短序列reads的拼接,并提供QUAST评估报告。
metaSPAdes 优点是拼接质量高,但对计算资源的要求较高。
megahit 优点是速度快、对计算资源要求低,但拼接质量不高。
命令:
(metawrap-env) [user@server ~]# metawrap assembly -1 read_qc/final_pure_reads_1.fastq -2 read_qc/final_pure_reads_2.fastq -o assembly_metaspades -m 600 -t 96 --metaspades
参数说明:
- -1 正向双末端测序fastq文件 (clean reads)
- -2 反向双末端测序fastq文件 (clean reads)
- -m 内存上限,内存消耗超过该设定时终止程序
- -t 计算线程数
- -l 重叠群contig长度下限 (default=1000)
- -o 结果输出目录
- --megahit # 用megahit拼接 (default)
- --metaspades # 用metaSPAdes拼接,取默认kmer=21,33,55
主要的输出结果包括:
- final_assembly.fasta 拼接的结果
- assembly_report.html quast报告
当选择较长的kmer时,对于丰度较高的物种基因组拼接有利;而当选择较短的kmer时,对于丰度较低的物种基因组有利。
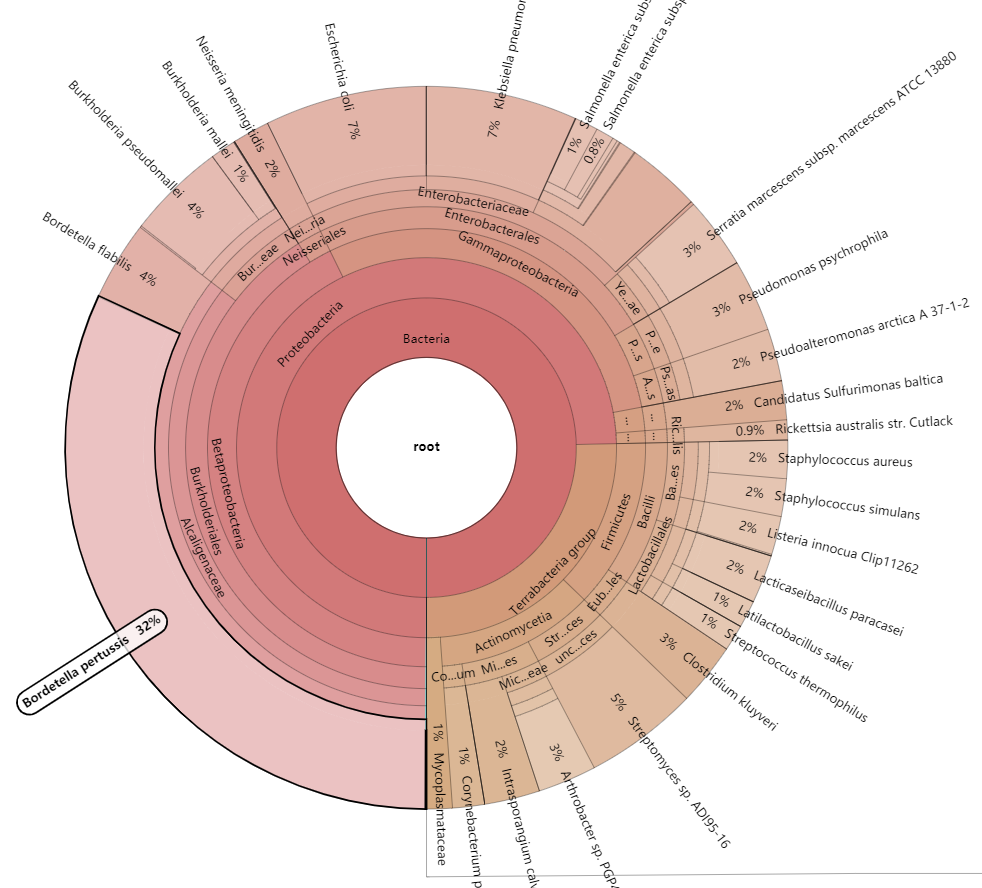
3.3 分类学注释 (kraken模块)
Kraken是一种快速而准确的分类学分类工具,它将reads与包含微生物独特kmers的数据库进行匹配。
(metawrap-env) [user@server ~]# metawrap kraken -o kraken -t 96 -s 1000000 assembly/final_assembly.fasta
参数说明: - -o 结果输出目录 - -t 计算线程数 - -s reads抽样数 (default=all) - --no-preload 禁止提前读取kraken数据库到内存 (程序运行将较慢, 但降低资源需求)
主要的输出结果包括:
- kronagram.html 注释结果html报告 (下图)
- final_assembly.kraken 每个contigs的注释结果
- final_assembly.krona 注释结果分类汇总
3.4 重叠群分箱 (binning模块)
metaWRAP binning借用了四个分箱工具(实为三个),分别是metabat2,concoct,maxbin2。
命令:
(metawrap-env) [user@server ~] # metawrap binning -o initial_binning -t 96 --metabat2 --maxbin2 --concoct -a assembly_megahit/final_assembly.fasta read_qc/final_pure_reads_*.fastq
参数说明:
- -a 拼接后的fasta文件
- -o 结果输出目录
- -t 计算线程数
- -m 内存使用量 (default=4)
- -l 分箱bin的长度下限 (default=1000bp). Note: metaBAT 的默认下限为1500bp
- --metabat2 用metaBAT2分箱
- --metabat1 用metaBAT分箱
- --maxbin2 用MaxBin2分箱
- --concoct 用CONCOCT分箱
- --universal 使用通用的标记基因 (将提升对古菌的分箱效果)
- --run-checkm 対分箱bin进行CheckM分析 (需要40GB+的内存)
- --single-end 非配对reads模式 (provide *.fastq files)
- --interleaved 输入reads文件中含有交叉存取的配对reads
主要输出结果:
- concoct_bins/ maxbin2_bins/ metabat2_bins/ 三个目录分别为三种软件的分箱结果
- work_files 分析过程中产生的过程文件
3.5 分箱提炼 (bin_refinement模块)
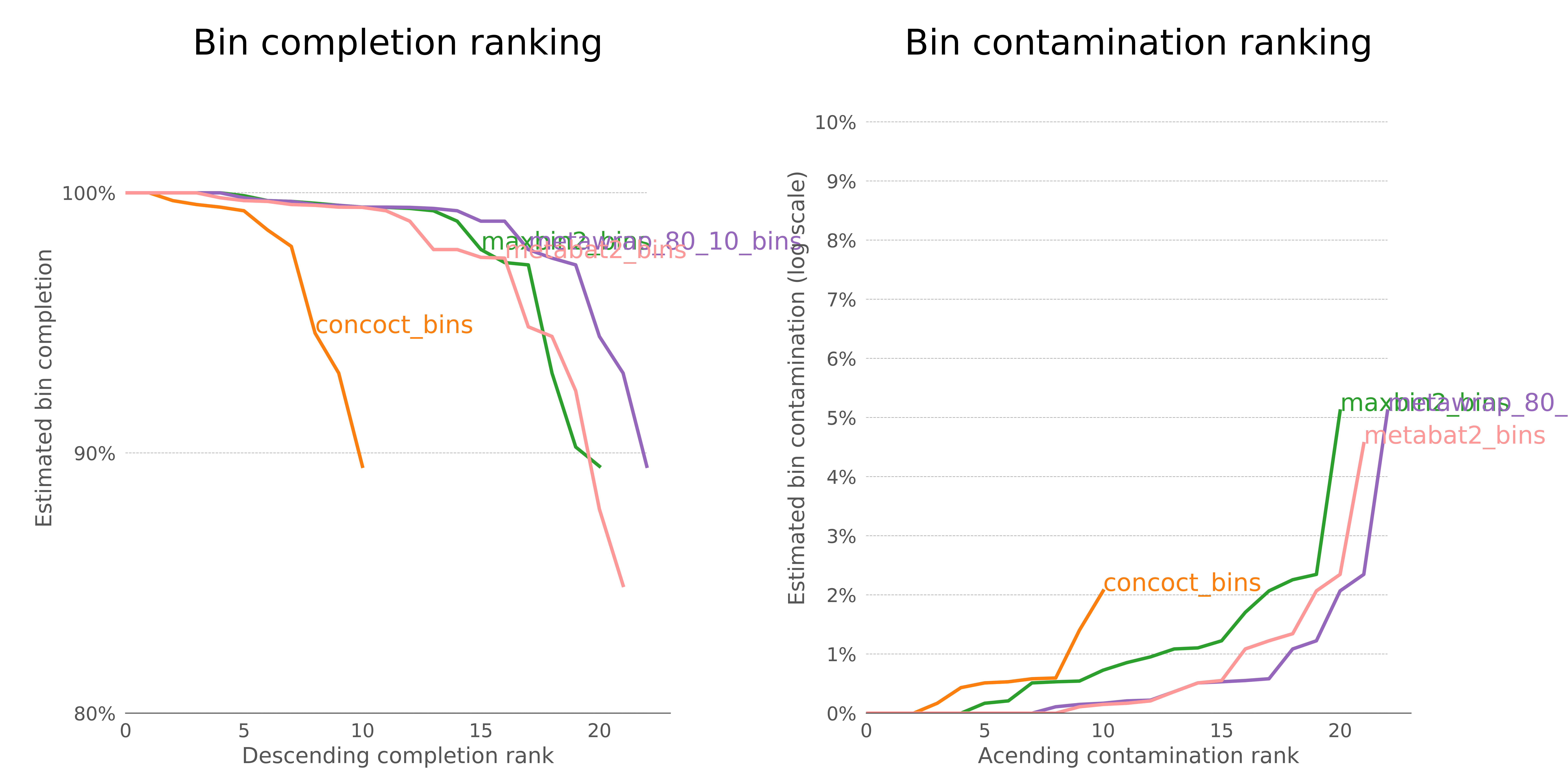
CheckM是一款对基因组质量进行评估的软件。其首先基于完整的已测序细菌基因组作为参考基因组,构建基因组的进化树,构建每个谱系(可以理解为一类物种)的单拷贝基因集(管家基因)。在使用时,将分箱bin与参考基因组一起建树,基于进化关系找到bin的参考物种,然后结合参考物种的单拷贝基因集,计算两个重要指标:完整度和污染度。获得每个bin的污染度、完整度信息后,然后通过bin_refinement模块挑选高质量的bins进行后续分析。
完整度 (Completeness):Bin基因与对应SCGs相比,基因数量是否完整,取值[0,100%],数值越大,表示Bin质量越好;
污染度 (Contamination):Bin基因包含多个物种的SCGs,即一个Bin存在多个物种的程度,取值[0,100%],数值越小,表示Bin质量越好。
命令:
(metawrap-env) [user@server ~] # metawrap bin_refinement -o bin_refinement -t 96 -A initial_binning/metabat2_bins/ -B initial_binning/maxbin2_bins/ -C initial_binning/concoct_bins/ -c 80 -x 10
参数说明:
- -o 结果输出目录
- -t 计算线程数
- -m 内存使用量 (default=40)
- -A / -B / -C 分别指向三种分箱结果
- -c 完整度阈值
- -x 污染度阙值
- --skip-refinement 不使用binning_refiner产生bin集合的交叉混合。如输入bin集合为A,B,C;binning_refiner将产生AB, BC, AC和ABC。
- --skip-checkm 跳过CheckM分析
- --skip-consolidation 选择提炼迭代中的最优版本
- --keep-ambiguous 当重叠群出现在不同的bin时,在各bin中保留 (default: 仅在最优bin中保留)
- --remove-ambiguous 当重叠群出现在不同的bin时,在各bin中都删除 (default: 仅在最优bin中保留)
- --quick 快速模式
结果目录bin_refinement中有三个原始bin的结果与统计:concoct_bins/ concoct_bins.stats concoct_bins.contigs; maxbin2_bins/ maxbin2_bins.stats maxbin2_bins.contigs;metabat2_bins/ metabat2_bins.stats metabat2_bins.contigs。metawrap_80_10_bins/ 目录包含最终分箱结果。
.stats文件包含每个bin的统计:完整度、污染度、GC含量、分类信息、N50、大小和来源。
(metawrap-env) [user@server ~] # head -n 3 metawrap_80_10_bins.stats
bin completeness contamination GC lineage N50 size binner
bin.21 100.0 0.0 0.295 Mycoplasma 430267 857632 binsA
bin.1 99.81 2.347 0.722 Streptomyces 350100 8215554 binsAC
提纯的结果在 bin_refinement/figures/ 目录中的图片,有eps图和png图。
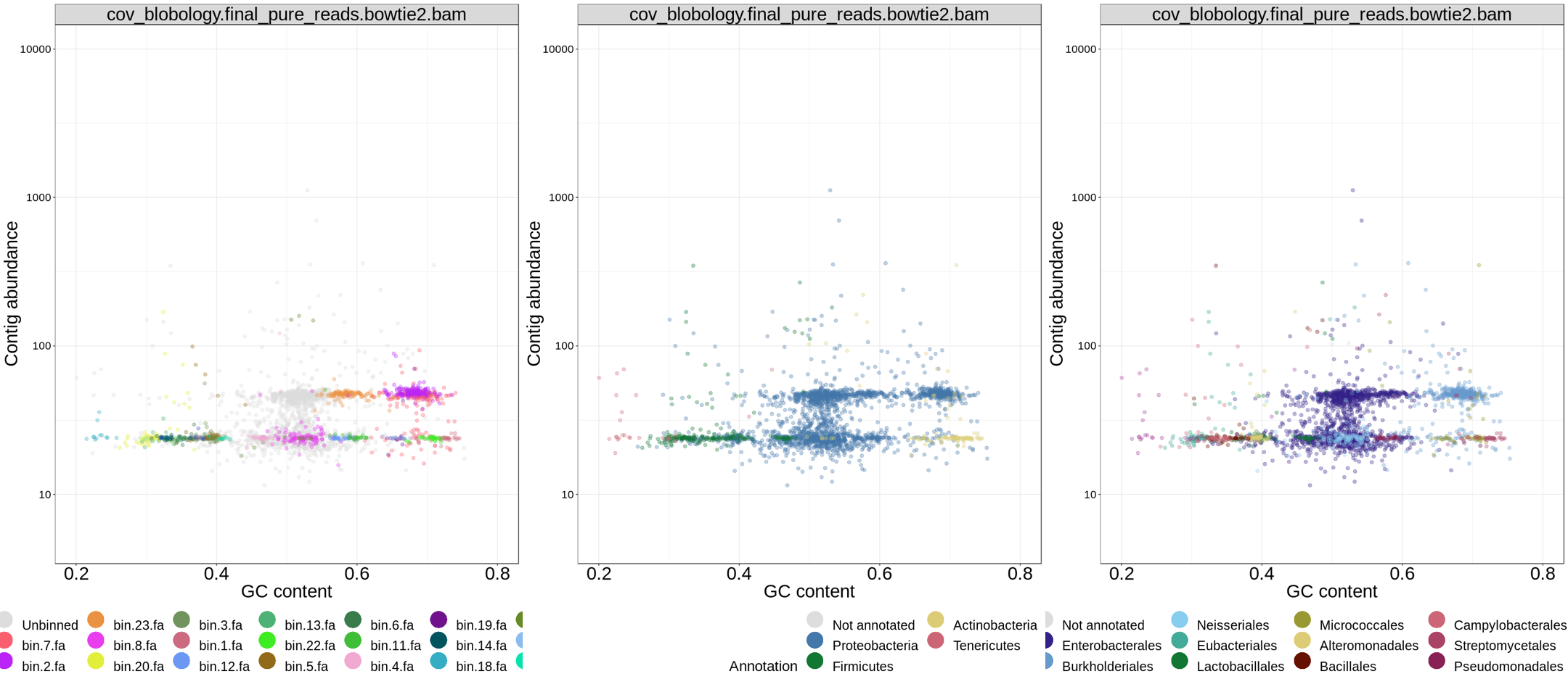
3.6 blobology
metawrap blobology模块,将contig的GC含量和丰度进行散点图可视化,并对不同分箱bin进行着色。
命令:
(metawrap-env) [user@server ~] # metawrap blobology -a assembly/final_assembly.fasta -o blobology -t 96 --bins bin_refinement/metawrap_80_10_bins read_qc/final_pure_reads_*.fastq
3.7 分箱丰度分析 (quant_bins模块)
metawrap quant_bins分箱丰度分析可以获取每个分箱bin在每个样品中的丰度。它依赖Salmon (用于转录组和宏基因组分析) 来实现定量,估计每一个重叠群contig的丰度,然后计算出bin在不同样本中的平均丰度。
命令:
(metawrap-env) [user@server ~] # metawrap quant_bins -b bin_refinement/metawrap_80_10_bins -t 96 -o quant_bins -a assembly/final_assembly.fasta read_qc/final_pure_reads_*.fastq
参数说明:
- -b 分箱bins的存放目录
- -o 结果输出目录
- -a 拼接后的fasta文件
- -t 计算线程数
主要输出结果:bin_abundance_table.tab,bins的丰度表。
由于本次分析所用的模拟数据仅有一组,相当于一个样本 (final_pure_reads_1.fastq和final_pure_reads_2.fastq),实际分析中如有多个样本,按顺序将不同样本的reads fastq文件写在命令之后即可。
3.8 分箱重组装 (reassemble_bins模块)
提纯之后的bin还可以通过重组装进一步改善结果。metawrap reaseemble_bins模块先调用bwa将原始reads比对到各个bins,然后进行重组装。当只有拼接结果得到提升时,才对结果进行更新。
命令:
(metawrap-env) [user@server ~] # metawrap reassemble_bins -o reassemble_bins -1 read_qc/final_pure_reads_1.fastq -2 read_qc/final_pure_reads_2.fastq -t 96 -m 600 -c 80 -x 10 -b bin_refinement/metawrap_80_10_bins
参数说明:
- -1 正向双末端测序fastq文件 (clean reads)
- -2 反向双末端测序fastq文件 (clean reads)
- -b bin_refinement 提纯之后bins所在目录
- -o 结果输出目录
- -t 计算线程数
- -m 内存使用量 (default=40)
- -c bin 完整度阙值
- -x bin 污染度阙值
- -l contig长度下限 (default=500)
- --strict-cut-off reads严格比对时允许的SNPs数上限 (default=2)
- --permissive-cut-off reads松弛比对时允许的SNPs数上限 (default=5)
- --skip-checkm 跳过CheckM分析
- --parallel 以并行方式运行spades,但每个线程仅针对一个bin
结果文件说明:
- original_bins 原始的bins
- original_bins.stats 原始bins的完整度、污染度、GC等统计信息
- reassembled_bins 重组装的bins
- reassembled_bins.stats 重组装的bins的完整度、污染度、GC等统计信息
此外还有,reassembly_results.png展示重新组装之后N50,完整度和污染度的变化。
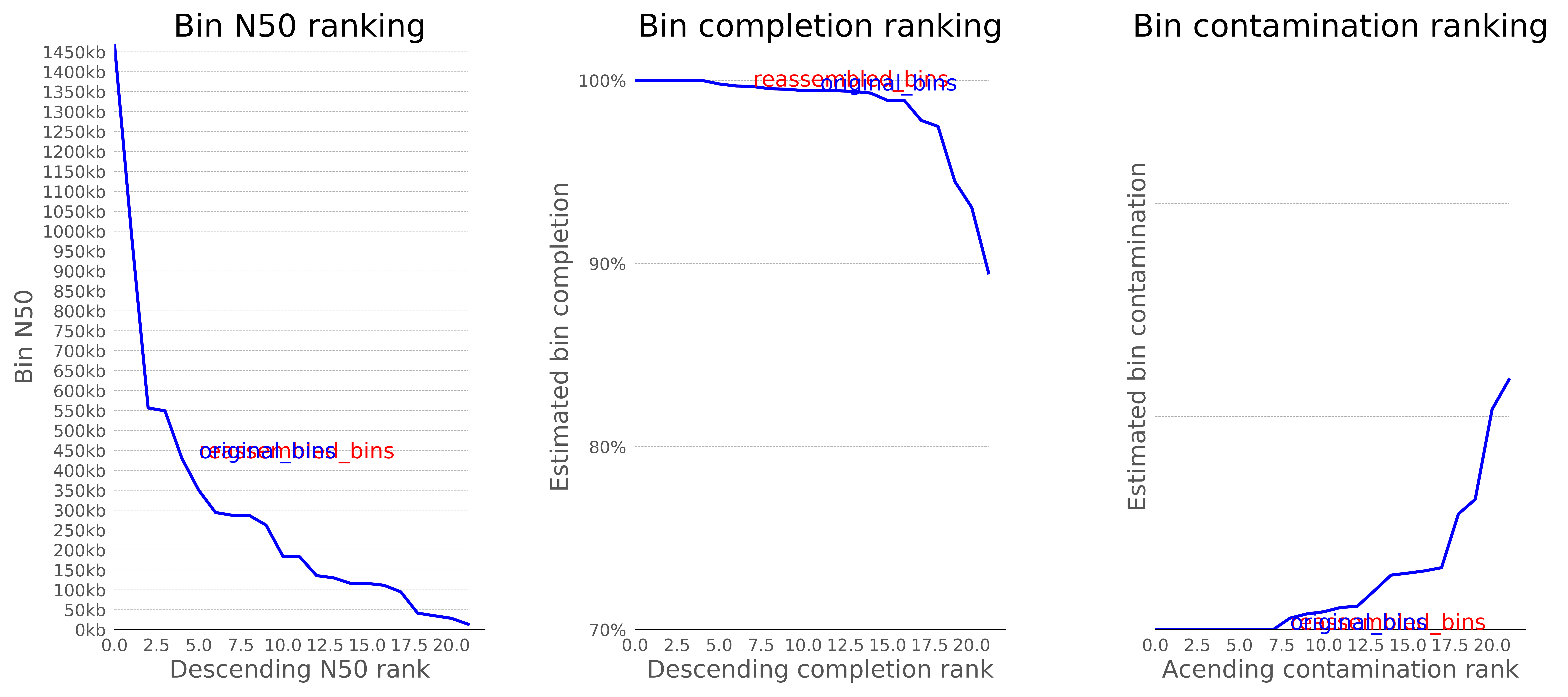
以及reassembly_bins.png展示CheckM对bin评估结果的可视化图。
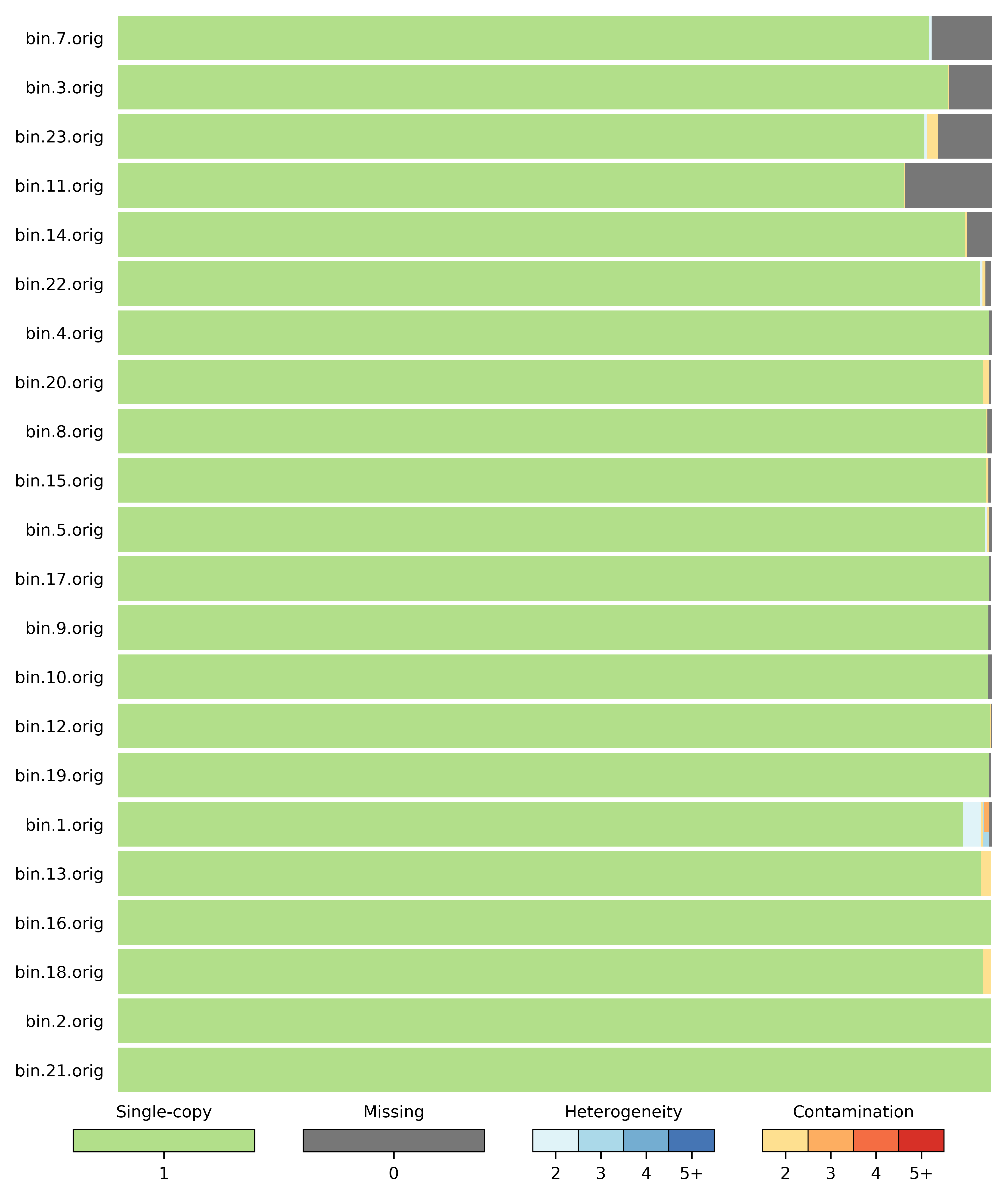
3.9 分箱分类学注释 (classify_bins模块)
metawrap classify_bins模块在NCBI_nt和NCBI_tax数据库基础上,使用工具Taxator-tk对每条contigs进行分类学注释,然后再估计bins的分类。注释结果的准确性也是由参考数据库决定的。
命令:
(metawrap-env) [user@server ~] # metawrap classify_bins -b reassemble_bin/reassembled_bins -o clssify_bin -t 8
结果文件说明:
(metawrap-env) [user@server ~] # head bin_taxonomy.tab
bin.20.orig.fa Bacteria;Firmicutes;Clostridia;Eubacteriales;Clostridiaceae;Clostridium;Clostridium kluyveri
bin.14.orig.fa Bacteria;Firmicutes;Bacilli;Bacillales;Staphylococcaceae
bin.7.orig.fa Bacteria;Proteobacteria;Betaproteobacteria;Burkholderiales;Burkholderiaceae;Burkholderia
bin.19.orig.fa Bacteria;Actinobacteria;Actinomycetia;Micrococcales;Micrococcaceae
bin.21.orig.fa Bacteria;Tenericutes;Mollicutes;Mycoplasmatales;Mycoplasmataceae;Mycoplasmopsis;Mycoplasma agalactiae
bin.3.orig.fa Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Pseudoalteromonadaceae
bin.5.orig.fa Bacteria;Firmicutes;Bacilli;Lactobacillales;Streptococcaceae;Streptococcus;Streptococcus thermophilus
bin.10.orig.fa Bacteria;Actinobacteria;Actinomycetia;Corynebacteriales;Corynebacteriaceae;Corynebacterium;Corynebacterium pseudotuberculosis
bin.13.orig.fa Bacteria;Proteobacteria;Epsilonproteobacteria;Campylobacterales;Thiovulaceae;Sulfurimonas;Candidatus Sulfurimonas baltica
bin.18.orig.fa Bacteria;Tenericutes;Mollicutes;Mycoplasmatales;Mycoplasmataceae;Mycoplasma;Mycoplasma neurolyticum
3.10 分箱基因功能注释 (annotate_bins模块)
metawrap annotate_bins利用Prokka进行基因功能注释。
命令:
(metawrap-env) [user@server ~] # metaWRAP annotate_bins -o annotate_bins -t 96 -b reassemble_bin/reassembled_bins/
结果文件说明:
- bin_funct_annotations prokka完成的gff文件
- bin_translatedgenes 翻译后的基因蛋白序列
- bin_untranslated_genes 未翻译的核苷酸序列
- prokka_out prokka原始输出结果
4. 参考文献
Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018 Sep 15;6(1):158. doi: 10.1186/s40168-018-0541-1.