
Snakemake: 搭建生信工作流程 (1)
![]()
Snakemake简介
Snakemake 是 Johannes Koster (也是Bioconda的作者) 为了数据分析能够更有效的重复,用 Python3 完成的一个工作流管理工具 (workflow management system)。因此继承了 Python 语言简单易读、逻辑清晰、便于维护的特点,同时它还支持 Python 语法。
Snakemake 工作流前后生成文件逻辑清晰,方便维护;而且可以无缝扩展到工作站、集群和云环境;作业调度控制方便;可以使用 conda 自动部署工作流所需要的软件环境。如果任务中断,当前的文件状态保留,已成功生成的文件不会被覆盖,已运行完的命令不会重复运行。
Snakemake 的基本组成单位叫 "rule",即"规则";每个 rule 里面又有多个 directives 指令(input、output、shell、run、threads等)。它的执行逻辑就是将各个 rule 利用 input -> output 连接起来,形成一个完整的工作流,当检测到 input,就执行相应 rule;检测到 output,就跳过相应 rule,根据这一规则,snakemake 还可以实现断点续接。
Snakemake 会将整个工作流程组织成有向无环图 (directed acyclic graph, DAG),并可视化 (借助dot命令)。
简单示例
Snakemake 官方文档以检测基因组突变为例,演示了其简单用法。该示例假设,对某基因组进行了 3 次二代重测序,拟通过bwa,samtools, bcftools 3 个第三方工具完成突变位点的检测。
前期准备
示例数据下载
Snakemake 官网提供了一套测试数据用于演示学习。
(base) user@server: # wget https://github.com/snakemake/snakemake-tutorial-data/archive/v5.24.1.tar.gz
(base) user@server: # tar --wildcards -xf v5.24.1.tar.gz --strip 1 "*/data" "*/environment.yaml"
在当前工作目录中会生成一个文件夹data和一个yaml文件environment.yaml (用于创建学习演示的 conda 环境)。
创建软件环境
通过mamba (一款 conda 加速器) 创建 conda 环境。
(base) user@server: # mamba env create --name snakemake-tutorial --file environment.yaml
初建工作流
首先进入snakemake-tutorial环境。
(base) user@server: # conda activate snakemake-tutorial
(snakemake-tutorial) user@server: # snakemake --help
然后利用文本编辑器 (vim) 创建并编辑文件,文件名varients_calling.snakefile。
Step 1. Generalizing the read mapping rule
官方示例中省略了建立bwa索引的操作 (示例数据
data中已有bwa比对所需的文件),直接从短序列比对开始。
在varients_calling.snakefile中首先写入一下内容:
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
注意 -Sb 之后的 - 与 重定向符号> 之间是有空格的。此时 - 代表的是通过管道传进来的数据位置。
关键词rule定义了snakemake的一个规则,名为 bwa_map。其中包含了 3 个 directives 指令:
- input 指定该 rule 需要的输入
- output 指定该 rule 将产生的输出
- shell 指定该 rule 将执行的 shell 命令
input 指定了2个输入,之间用,分隔。这两个输入文件实际上是 shell 所指定的命令bwa mem所需要的。在bwa mem命令行中{input}指代了 input 所指定的两个输入文件。因此可以理解一对花括号 {} 起到了 wildcards 通配符的功能。所以在第二个输入中{sample}指代了所有在data/samples/目录下的fastq文件。
这里当用{input}指代两个输入时,实际上相当于字符串的串联,其结果为data/genome.fa data/samples/{sample}.fastq。而data/samples/目录中又有 3 个fastq文件,所以{input}相当于:
data/genome.fa data/samples/A.fastqdata/genome.fa data/samples/B.fastqdata/genome.fa data/samples/C.fastq
它们之间是逻辑或关系,也就意味着bwa mem命令将要运行 3 次,分别针对 3 个fastq文件进行基因组比对。
output 指令规定了bwa mem命令生成的 (实际上是其结果通过|管道交给了samtools view -Sb) bam文件将会存放至mapped_reads/目录中,该目录在不存在时会自动建立。
至于bwa mem命令的执行,以及计算资源的调用,则可完全交给 snakemake 来处理。
首先执行如下命令:
(snakemake-tutorial) user@server: # snakemake --dry-run --printshellcmds --snakefile varients_calling.snakefile mapped_reads/A.bam
- --dry-run or -n 只显示执行计划,而不是实际执行这些步骤
- --printshellcmds or -p 将每个rule所执行的命令输出到屏幕
- --snakefile 工作流文件,如不指定snakemake会在工作目录中寻找名为
Snakefile的文件 - [target [target ...]] 目标文件 (必须的),即mapped_reads/A.bam
Building DAG of jobs...
Job counts:
count jobs
1 bwa_map
1
[Fri May 14 13:33:48 2021]
rule bwa_map:
input: data/genome.fa, data/samples/A.fastq
output: mapped_reads/A.bam
jobid: 0
wildcards: sample=A
bwa mem data/genome.fa data/samples/A.fastq | samtools view -Sb - > mapped_reads/A.bam
Job counts:
count jobs
1 bwa_map
1
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
从屏幕输出可以看出,由于目标文件仅指定了mapped_reads/A.bam,所以该工作流的任务 job 只有 1 个 rule即bwa_map,且该rule只执行 1 次。再看下例:
(snakemake-tutorial) user@server: # snakemake -n -p --snakefile varients_calling.snakefile mapped_reads/{A,B,C}.bam
Building DAG of jobs...
Job counts:
count jobs
3 bwa_map
3
[Fri May 14 13:38:59 2021]
rule bwa_map:
input: data/genome.fa, data/samples/B.fastq
output: mapped_reads/B.bam
jobid: 0
wildcards: sample=B
bwa mem data/genome.fa data/samples/B.fastq | samtools view -Sb - > mapped_reads/B.bam
[Fri May 14 13:38:59 2021]
rule bwa_map:
input: data/genome.fa, data/samples/C.fastq
output: mapped_reads/C.bam
jobid: 2
wildcards: sample=C
bwa mem data/genome.fa data/samples/C.fastq | samtools view -Sb - > mapped_reads/C.bam
[Fri May 14 13:38:59 2021]
rule bwa_map:
input: data/genome.fa, data/samples/A.fastq
output: mapped_reads/A.bam
jobid: 1
wildcards: sample=A
bwa mem data/genome.fa data/samples/A.fastq | samtools view -Sb - > mapped_reads/A.bam
Job counts:
count jobs
3 bwa_map
3
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
命令行通配符{}给了 3 个目标文件,这时bwa_map则要被执行 3 次,分别针对 3 个目标文件。注意:执行的顺序是随机的!
Step 2. Sorting read alignments
完成短序列 reads 比对后,需要对 bam 结果进行排序。因此,应继续向工作流中添加新的任务。编辑varients_calling.snakefile,追加一下内容:
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.sorted.bam"
shell:
"samtools sort -O bam -o {output} {input}"
此时 input 为上一个 rule 的 output,snakemake 会自动建立sorted_reads文件夹。
执行如下命令:
(snakemake-tutorial) user@server: # snakemake -n -p --snakefile varients_calling.snakefile sorted_reads/A.sorted.bam
Building DAG of jobs...
Job counts:
count jobs
1 bwa_map
1 samtools_sort
2
[Fri May 14 17:06:34 2021]
rule bwa_map:
input: data/genome.fa, data/samples/A.fastq
output: mapped_reads/A.bam
jobid: 1
wildcards: sample=A
bwa mem data/genome.fa data/samples/A.fastq | samtools view -Sb - > mapped_reads/A.bam
[Fri May 14 17:06:34 2021]
rule samtools_sort:
input: mapped_reads/A.bam
output: sorted_reads/A.sorted.bam
jobid: 0
wildcards: sample=A
samtools sort -f mapped_reads/A.bam sorted_reads/A.sorted.bam
Job counts:
count jobs
1 bwa_map
1 samtools_sort
2
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
这时指定了一个目标文件sorted_reads/A.sorted.bam,工作流的任务 job 变为 2 个 rule 即 bwa_map和samtools_sort,所以针对产生目标文件sorted_reads/A.sorted.bam将要执行 2 个操作。
当同样使用通配符sorted_reads/{A,B,C}.sorted.bam,涵盖所有数据时,屏幕输出内容会比较长,可用--quiet参数来简化输出:
(snakemake-tutorial) user@server: # snakemake -np --quiet --snakefile varients_calling.snakefile sorted_reads/{A,B,C}.sorted.bam
Job counts:
count jobs
3 bwa_map
3 samtools_sort
6
bwa_map和samtools_sort将针对 3 个序列文件,分别执行 3 次。
Step 3. Indexing read alignments
完成比对结果的排序,利用samtools index建立索引。追加一个新的 rule 至 varients_calling.snakefile。
rule samtools_index:
input:
"sorted_reads/{sample}.sorted.bam"
output:
"sorted_reads/{sample}.sorted.bam.bai"
shell:
"samtools index {input}"
(snakemake-tutorial) user@server: # snakemake -np --quiet --snakefile varients_calling.snakefile sorted_reads/{A,B,C}.sorted.bam.bai
Job counts:
count jobs
3 bwa_map
3 samtools_sort
3 samtools_index
9
注意这里的目标文件是sorted_reads/{A,B,C}.sorted.bam.bai。至此,工作流中已经添加了 3 个功能的 rules,分别完成"序列比对","比对结果排序"和"排序结果的索引"。
仔细观察会发现,以上 3 个 rules 的排序并非工作流的逻辑顺序,而实际上是 3 个 rules 名称的字母顺序。
要获得工作流的逻辑顺序,snakemake提供了一种更直观的方式,即生成DAG图。
(snakemake-tutorial) user@server: # snakemake --dag --snakefile varients_calling.snakefile sorted_reads/{A,B,C}.sorted.bam.bai | dot -Tsvg > dag.svg
借助--dag参数,并将结果通过|管道符传给dot命令,可生成一个svg格式的DAG图 (下图)。
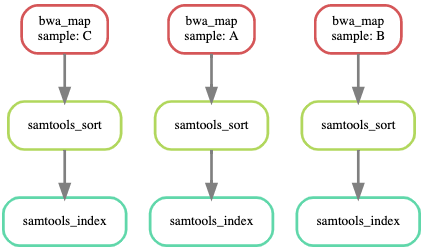
对排序的bam建立索引得到bai文件,对于下一步变异检测不是必须的,不知snakemake官方文档为何要设计这一步。
Step 4. Calling genomic variants
接下来即可进行突变位点的检测,该分析需要借助bcftools中的两个子命令bcftools mpileup和bcftools call。
官方文档用的是
samtools mpileup。
首先需要用bcftools mpileup 将在所有排序且已索引的bam文件,以及基因组文件 data/genome.fa的基础上,生成 bcf 文件;再通过|管道将 bcf 文件传给bcftools call,将变异信息以 vcf 文件形式输出。
再追加一个新的 rule 至 varients_calling.snakefile:
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.sorted.bam", sample=['A', 'B', 'C']),
bai=expand("sorted_reads/{sample}.sorted.bam.bai", sample=['A', 'B', 'C'])
output:
"calls/all.vcf"
shell:
"bcftools mpileup -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
input.bai 非必需,为了生成与官方一致的DAG图,特保留。
bcftools mpileup所需要的排序且已索引的bam文件和基因组文件 data/genome.fa,分别由 input 的fa和bam定义。
其中fa是一个固定文件,而bam是由通配符{sample}表示的多个文件。前面 rules 的分阶段执行snakemake的时,需要指定目标文件,例如:
# snakemake -np --quiet --snakefile varients_calling.snakefile sorted_reads/{A,B,C}.sorted.bam.bai
sorted_reads/{A,B,C}.sorted.bam.bai 传到 varients_calling.snakefile 实际上是由 rule samtools_index 的 output sorted_reads/{sample}.sorted.bam.bai 接收的,所以{sample} 等于 {A,B,C} 。
按照执行snakemake的要求,当要执行 rule bcftools_call 时,需要的目标文件应该为calls/all.vcf。这时问题出现了,因为此时没有外部 {sample} 向varients_calling.snakefile传入 {A,B,C}了。为解决这一问题,snakemake设计了expand。
expand的用法,形式上类似一个 Python 函数,接入一个带有通配符的字符串,然后再以列表的形式定义通配符所指代的对象。该实例中 expand("sorted_reads/{sample}.sorted.bam", sample=['A', 'B', 'C']),等价于:
['sorted_reads/A.sorted.bam', 'sorted_reads/B.sorted.bam', 'sorted_reads/C.sorted.bam']
该函数尤其在处理多个通配符时,作用则更加明显,如:
expand("sorted_reads/{sample}.{replicate}.bam", sample=['A', 'B'], replicate=[0, 1])
等同于:
["sorted_reads/A.0.bam", "sorted_reads/A.1.bam", "sorted_reads/B.0.bam", "sorted_reads/B.1.bam"]
另外,当有多个 input 时,在 shell 的使用方式是在input后加.并跟上不同的input的具体名称,例如:input.fa和input.bam。
到此为止,突变检测的工作流以基本完成,最后再生成一个DAG图,纵览工作流的逻辑过程如下:
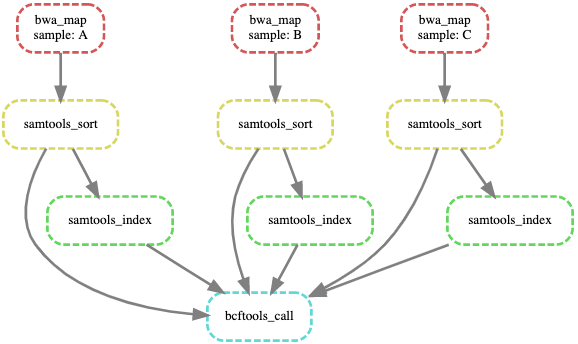
如在rule bcftools中不设定input.bai,以上DAG图中,则不再有 3 个samtools_index节点,以及与之相连的边。
隐藏目标文件
以上示例中,执行snakemake都需要指定目标文件,除了目标文件名外,snakemake还支持 rule 的名作为目标。
追加一个 rule 至 varients_calling.snakefile:
rule all:
input:
"calls/all.vcf"
这个 rule 只定义了一个input,即 rule bcftools_call 的 output。这时即可通过一下方式执行该工作流:
(snakemake-tutorial) user@server: # snakemake --cores 6 --snakefile varients_calling.snakefile all
这里不再通过-np参数只显示执行计划,而是分配 6 个线程,来运行varients_calling.snakefile所定义的完整工作流了 (也就可以理解最后一个rule取名all的意义了)。
当在运行snakemake时不指定目标,snakemake会默认执行第一个规则,因此如果将 rule all 作为Snakefile的第一个rule,则可在执行时省略目标。
如
--cores之后不跟有设定数值,snakemake则会使用全部线程。