
Nanopore三代测序数据拼接——Trycycler

Trycycler简介
Trycycler是一种三代测序结果拼接工具,它可以将多个不同Nanopore/PacBio三代测序拼接软件产生的组装结果进行整合,进而产生一个最佳组装效果的一致基因组序列。
Trycycler在Linux和macOS上运行需要python3.6版本或者更高,其还需要python包如(Edlib, NumPy and SciPy),此外还需要外部工具包如Minisam、Minimap2、Mash、Muscle以及你想使用的基因组组装工具,在运行前因检查是否安装。
Trycycler的安装可通过conda、git、pip安装,具体安装过程点击此处。
Trycycler的用法
Step 0: 序列质控
测序仪器产出原始fast5文件经过basecalling (Nanopore Guppy等),输出fastq文件。在进行组装之前,我们利用fitlong软件来进行质控。
(genomics) user@server:~ # filtlong --min_length 1000 --keep_percent 95 input_reads.fastq.gz > reads.fastq #此处推荐用质量进行质控,保留95%数据
Basecalling一般由测序公司完成
Step 1: 对clean reads进行抽样
为了给不同的第三方拼接软件,准备拼接用的数据,Trycycler从原clean reads集 (上一步质控输出的reads.fastq文件)中进行随机抽样产生多组子数据集。
(genomics) user@server:~ # trycycler subsample --count 12 --min_read_depth 25 --reads reads.fastq --out_dir read_subsets #生成的结果在read_subsets/sample_*.fastq默认有12个
参数说明:
- --reads 输入fastq文件
- --out_dir 输出文件夹
- --count 产生的子数据集个数 (default: 12)
- --min_read_depth 最小read覆盖深度 (default: 25.0)
- --genome_size 基因组大小估计值 (default: auto)
- --threads 计算线程数 (default: 16)
Step 2: 序列组装
利用不同的基因组组装软件如Canu、flye、raven (便于命令行展示) 对12个子数据集进行组装。
(genomics) user@server:~ # flye --nano-raw read_subsets/sample_01.fastq --threads 64 --plasmids --out-dir assembly_01
(genomics) user@server:~ # canu -p assembly_02 -d assembly_02 genomeSize=<number>[g|m|k] -nanopore-raw read_subsets/sample_02.fastq
(genomics) user@server:~ # raven --threads 64 read_subsets/sample_03.fastq > assemblies/assembly_03.fasta
最终将拼接结果集中存放至assemblies文件夹,并分别命名为assembly_00.fasta~assembly_11.fasta等12个组装好的基因组文件。
12组子数据集,3个拼接软件可分别分配4个数据集。
第三方拼接软件推荐:Canu,flye,necat
Step 3: 初步拼接结果的聚类分析
针对所有子数据集合的拼接结果 (基于不同的软件),即产生的contigs,进行基于contigs间Mash距离的聚类分析 (complete-linkage clustering)。
(genomics) user@server:~ # trycycler cluster --assemblies assemblies/*.fasta --reads reads.fastq --out_dir trycycler
参数说明:
- --assemblies 输入的拼接结果 (子数据集)
- --reads 输入fastq文件
- --out_dir 输出文件夹
- --min_contig_len contig长度下限 (default: 1000)
- --min_contig_depth 相对contig深度下限 (相对于平均contig深度,default: 0.1)
- --distance Mash距离阈值 (default: 0.01)
- --threads 计算线程数 (default: 16)
在输出的trycycler目录将产生以下文件/文件夹:
- cluster_001、cluster_002等目录。每个聚类簇都有一个目录,且在这些目录中都包含一个名为1_contigs的子目录,存放了属于该聚类簇的所有contigs文件 (fasta格式)。
- contigs.phylip:PHYLIP格式中所有contigs之间的Mash距离的矩阵。
- contigs.newick:从距离矩阵构建的contigs的FastME树。可用进化树编辑软件,如FigTree显示。
聚类分析的目的是得到利用不同拼接软件的结果中一致的contigs,同时发现潜在的错误拼接的contigs,这些错误拼接的contigs多为某单个软件的产物,在聚类树上往往形成单独的分枝。
可通过查看的contigs.newick树文件来手动选择好的集群,删除或从1_contigs中移除那些不好的不能聚类的contigs。
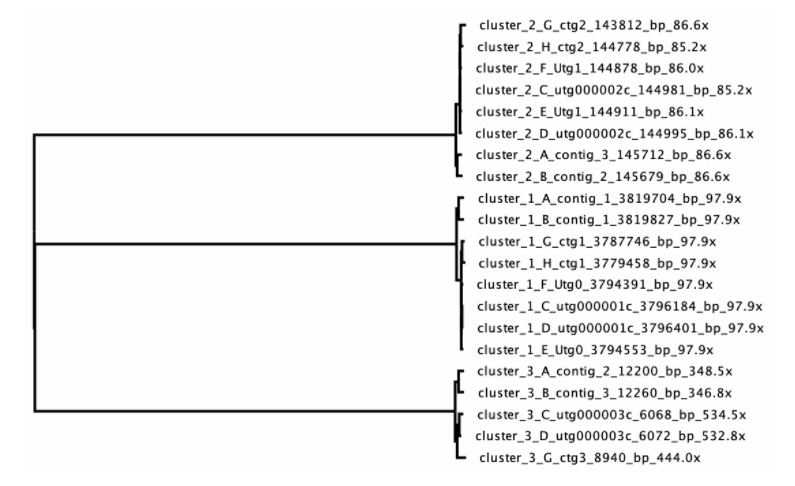
在下图所示的例子中,所有的contigs都能较好的聚类。且从contig长度可以看出cluster_1可能为染色体,cluster_2和cluster_3分别为质粒。但cluster_3中的contigs长度差异较大,需要在下一步中进行优化。
而在下图所示的例子中,cluster_1和cluster_3聚类效果较好,cluster_2 (可能为质粒) 看起来也还可以,但cluster_4和cluster_6和其紧密相关,但分别只有一个contigs,估计为错误组装的cluster_2,或为cluster_2的片段,可排除。cluster_5与其它聚类簇都相距较远,且深度较低 (17.8倍),也可排除。
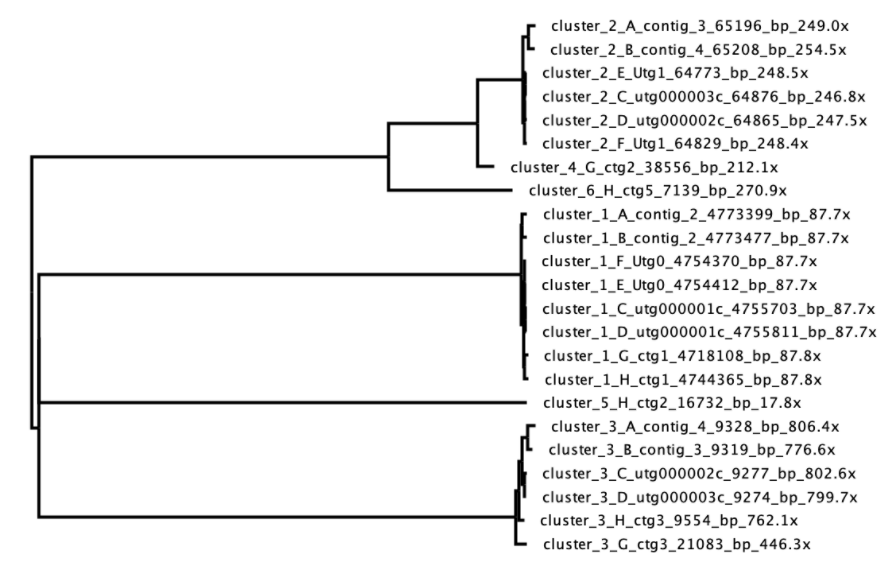
该步是Trycycler软件操作中唯一需要人工判断的。也是拼接结果质量的关键。关于此部分的更多例子可点击此处。
Step 4: 调整Contigs
对各个聚类簇中的contigs进行调整。首先进行initial check,以保证所有contigs都足够相似 (相对长度足够均匀,Mash距离足够小);然后保证所有contigs的链方向一致;再者如果该复制子为环状,排除所有影响成环的问题,并将所有的contigs的起始位置保持一致;最后再进行final alignment check,让所有标准化/成环化的contigs尽可能的相似,以便进行下一步的多序列比对。
如果顺利的话,程序可以自己完成,否则需要手动修改某些参数。
(genomics) user@server:~ # trycycler reconcile --reads reads.fastq --cluster_dir trycycler/cluster_001
(genomics) user@server:~ # trycycler reconcile --reads reads.fastq --cluster_dir trycycler/cluster_002
(genomics) user@server:~ # trycycler reconcile --reads reads.fastq --cluster_dir trycycler/cluster_003
参数说明:
- --reads 输入fastq文件
- --linear 当基因组为非环状基因组时需要
- --cluster_dir 聚类分析的聚类簇对应的文件夹
- --threads 计算线程数
- --max_mash_dist contigs间mash距离的上限 (default = 0.02)
- --max_length_diff contigs间长度偏差上限 (default = 1.1,没有contigs可以任意其它contigs长10%)
- --max_add_seq 为环化contig,允许延长序列的长度 (default = 1000,最长延长1000bp)
- --max_add_seq_percent 为环化contig,允许延长序列的百分比 (default = 5,最长延长contig长度的5%)
- --max_trim_seq 为环化contig,允许剪切序列的长度 (default = 50000 ,最长剪切50000bp)
- --max_trim_seq_percent 为环化contig,允许剪切序列的百分比 (default = 10,最长剪切contig长度的10%)
- --min_identity contigs间两两全局比对的一致性下限 (default = 98)
- --max_indel_size contigs间两两全局比对的插入缺失数上限 (default = 250)
每个cluster需要分别进行contigs调和。
Step 5: 多序列比对
此步为多序列比对,通过以下命令运行
(genomics) user@server:~ # trycycler msa --cluster_dir trycycler/cluster_001
(genomics) user@server:~ # trycycler msa --cluster_dir trycycler/cluster_002
(genomics) user@server:~ # trycycler msa --cluster_dir trycycler/cluster_003
参数说明:
- --cluster_dir 聚类分析的聚类簇对应的文件夹
- --kmer 序列分割的kmer长度 (default = 32)
- --step 序列分割的步长 (default = 1000)
- --lookahead 序列分割的先导范围 (default = 10000)
- --threads 计算线程数
关于序列分割的参数不需要调整,默认即可。
Step 6: 分解clean Reads
根据每个contigs聚类簇,将clean reads分配到各个聚类簇。
(genomics) user@server:~ # trycycler partition --reads reads.fastq --cluster_dirs trycycler/cluster_*
参数说明:
- --reads 输入fastq文件
- --cluster_dir 聚类分析的聚类簇对应的文件夹 (可用
*通配符) - --min_aligned_len read匹配长度的下限 (default = 1000)
- --min_read_cov read匹配区比例 (coverage) 的下限 (default = 90.0)
- --threads 计算线程数
Step 7: 生成一致序列
根据每个聚类簇中的contigs比对结果,生成一个一致序列。
(genomics) user@server:~ # trycycler consensus --cluster_dir trycycler/cluster_001
(genomics) user@server:~ # trycycler consensus --cluster_dir trycycler/cluster_002
(genomics) user@server:~ # trycycler consensus --cluster_dir trycycler/cluster_003
参数说明:
- --cluster_dir 聚类分析的聚类簇对应的文件夹
- --linear 当基因组为非环状基因组时需要
- --min_aligned_len read匹配长度的下限 (default = 1000)
- --min_read_cov read匹配区比例 (coverage) 的下限 (default = 90.0)
- --threads 计算线程数
生成的结果在 trycycler/cluster_*/7_final_consensus.fasta
Step 8: 打磨拼接结果
上一步产生的一致性序列即是最后的拼接结果,为了进一步的提高拼接结果的准确度,对于Nanopore的测序数据还可以用Medaka来“打磨”一致性序列。
Medaka需要指定针对不同测序设备的特定模型。
如果除了三代数据,还有Illumina二代数据的话,可以使用Pilon 来“打磨”一致性序列。
An important caveat (read it carefully)
Trycycler does not ensure a perfect assembly of the underlying genome, because systematic basecalling errors can create small-scale sequence errors. Incorrect homopolymer lengths are a common example of this problem, e.g. AAAAAAAA becoming AAAAAAA (read more here).
But if all goes well when running Trycycler, small-scale errors will be the only type of error in its consensus long-read assembly. You can then polish your Trycycler assembly to repair these small-scale errors, e.g. long-read polishing with Medaka then short-read polishing with Pilon. A Trycycler+Medaka+Pilon approach to assembly can therefore yield the best possible bacterial genome: Trycycler fixes the medium-to-large-scale errors while Medaka and Pilon fix the small-scale errors.
图片摘自:Trycycler