SRA数据库的使用
sra数据库
一:简介
SRA ( Sequence Read Archive)是NCBI旗下的数据库之一,其作用是存储包括Illumina、454、IonTorrent、Complete Genomic、PacBio和Oxford Nanpores在内的二/三代测序技术所产生的原始序列数据。这些数据可以提交给GeneBank(美国)、EMBL(欧洲)和DDBJ(日本)这三大核酸数据库之一,并会在三者间共享,这三大核算数据库组成的联合核苷酸数据库被称为INSDC(国际核苷序列联合数据库)。除了原始序列数据外,SRA现在也存在raw reads在参考基因的比对信息。
SRA的官方网址为:https://www.ncbi.nlm.nih.gov/sra/
根据SRA数据产生的特点,将SRA数据分为四类:
1. Studies-- 研究课题
2. Experiments-- 实验设计
3. Runs-- 测序结果集
4. Samples-- 样品信息
SRA中数据结构的层次关系为:Studies->Experiments->Samples->Runs.
1. Studies是就实验目标而言的,一个study 可能包含多个Experiment。
2. Experiments包含了Sample、DNA source、测序平台、数据处理等信息。
3. 一个Experiment可能包含一个或多个runs。
4. Runs 表示测序仪运行所产生的reads。
SRA数据库用不同的前缀加以区分:
1. SRP,ERP,DRP表示Studies;
2. SRX,ERX,DRX 表示 Experiments;
3. SRS,ERS,DRS 表示 Samples;
4. SRR ,ERR,DRR表示 Runs;
二:SRA界面详解
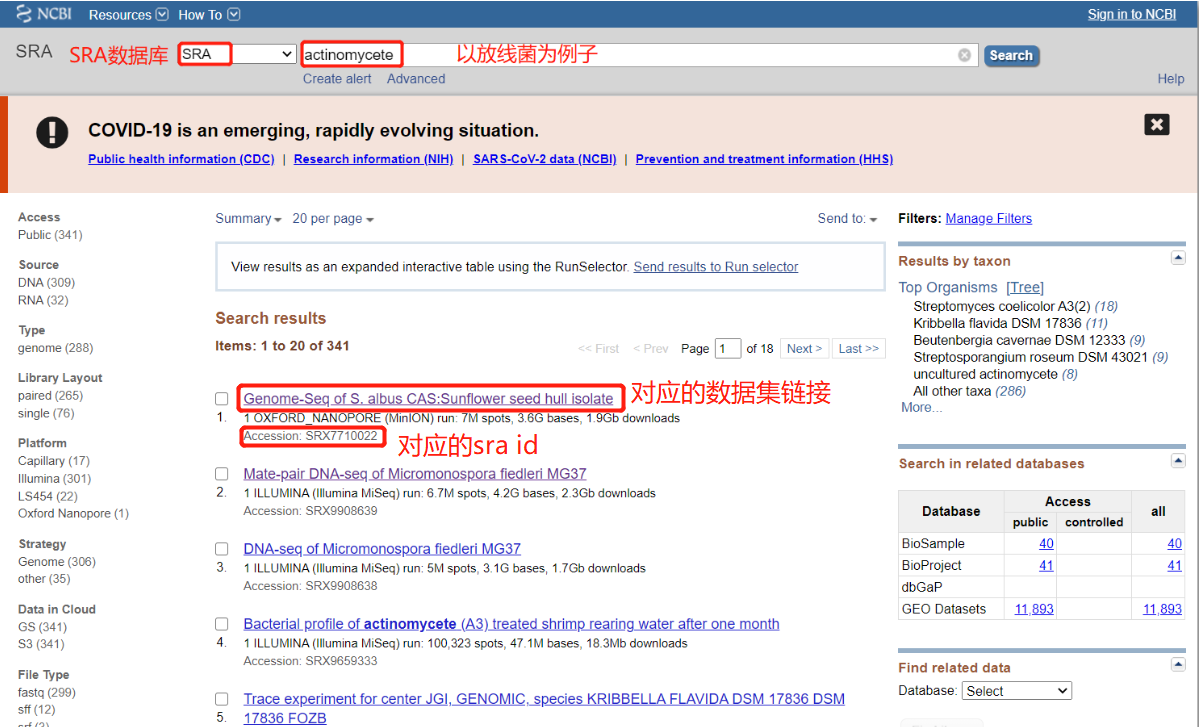
1.1 以放线菌为例进行搜索。
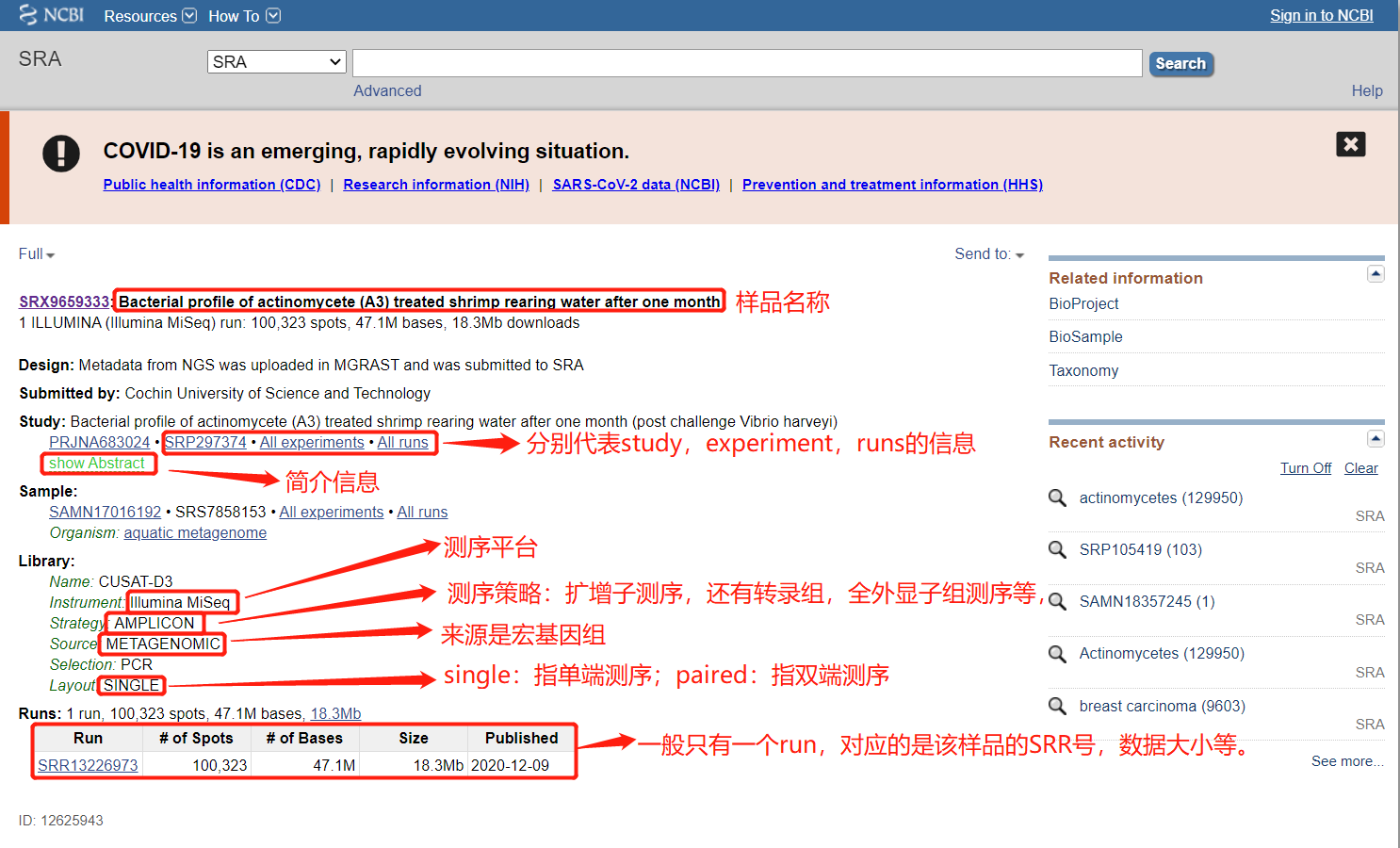
1.2 点击进去其中一个项目
2. 也可以用具体的sra id 号进行搜索
直接在搜索框输入sra id号就可以了
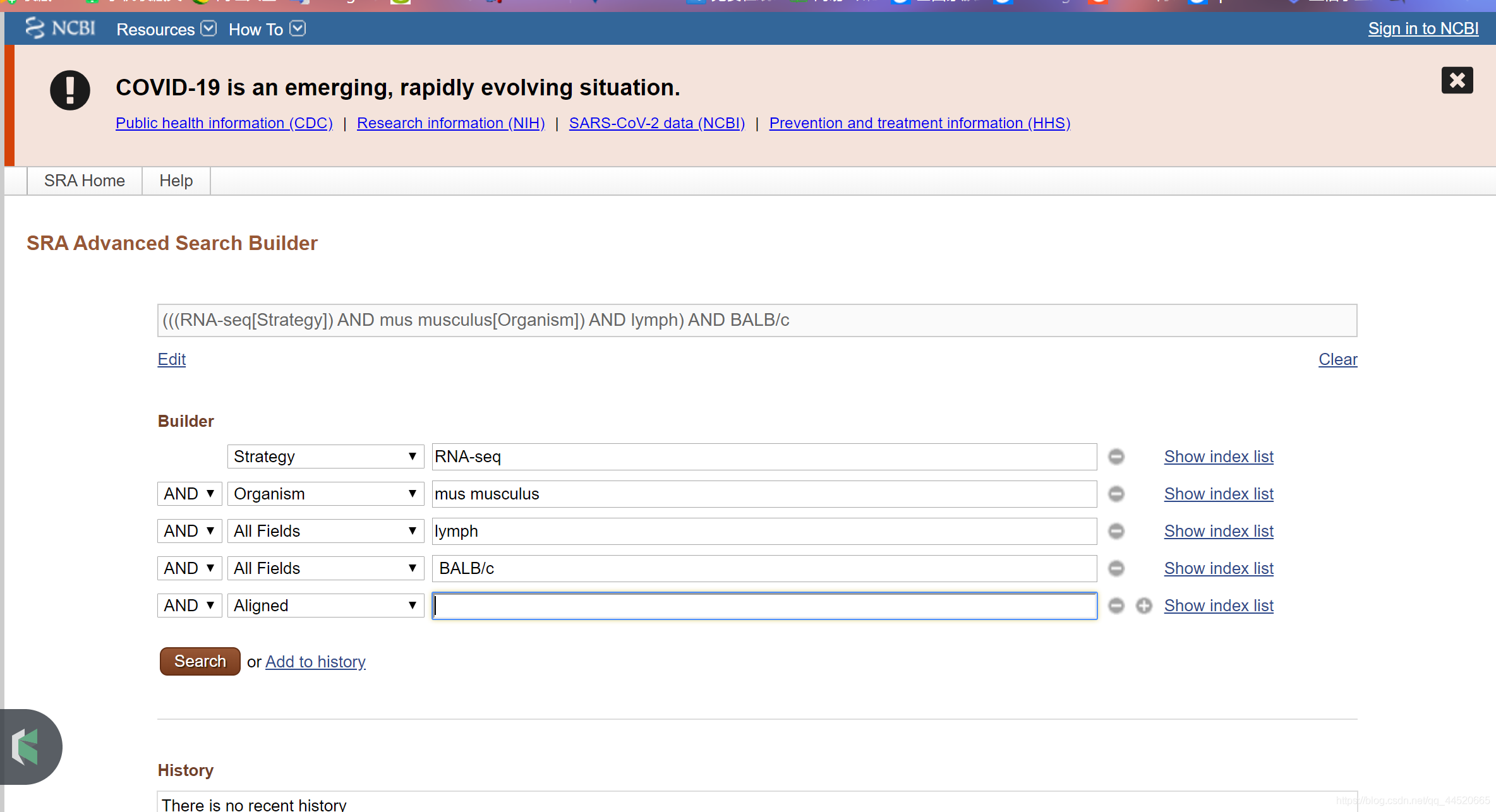
3. SRA的高级搜索
以BALB / c小鼠淋巴结组织的RNA-Seq记录为例:
三:数据下载
一般我们使用NCBI提供的SRA Toolkit来下载数据,下面是SRA Toolkit用法
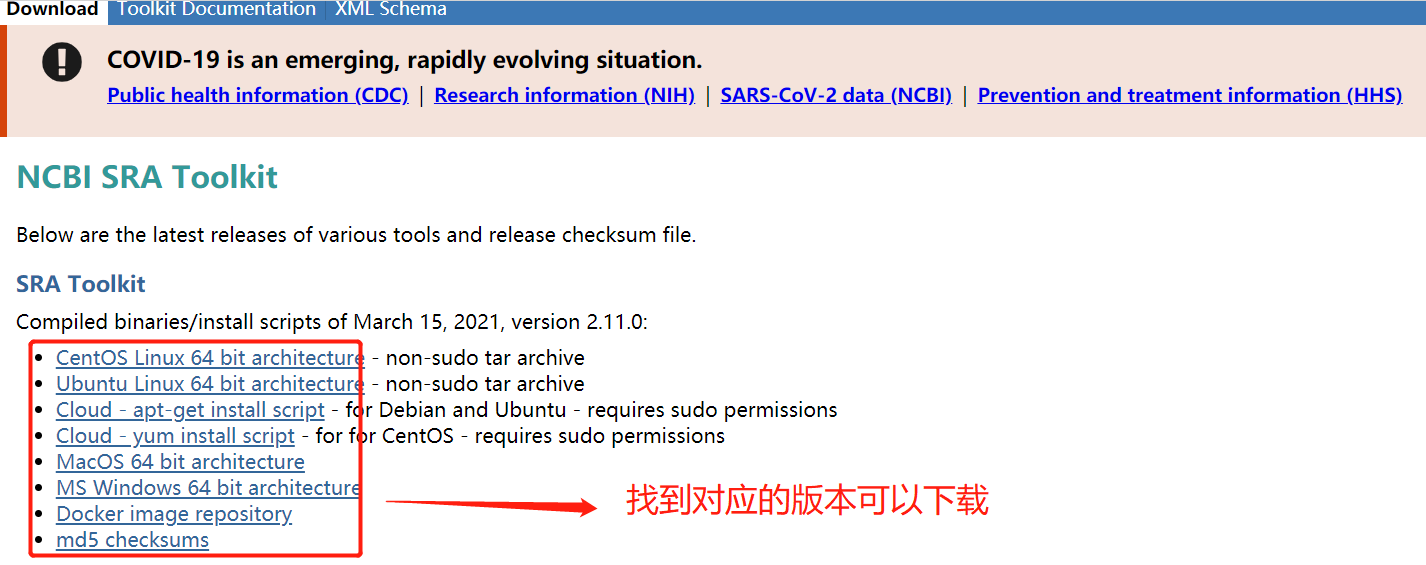
1. windows下安装
cd + 安装目录(一直到可执行程序bin下)
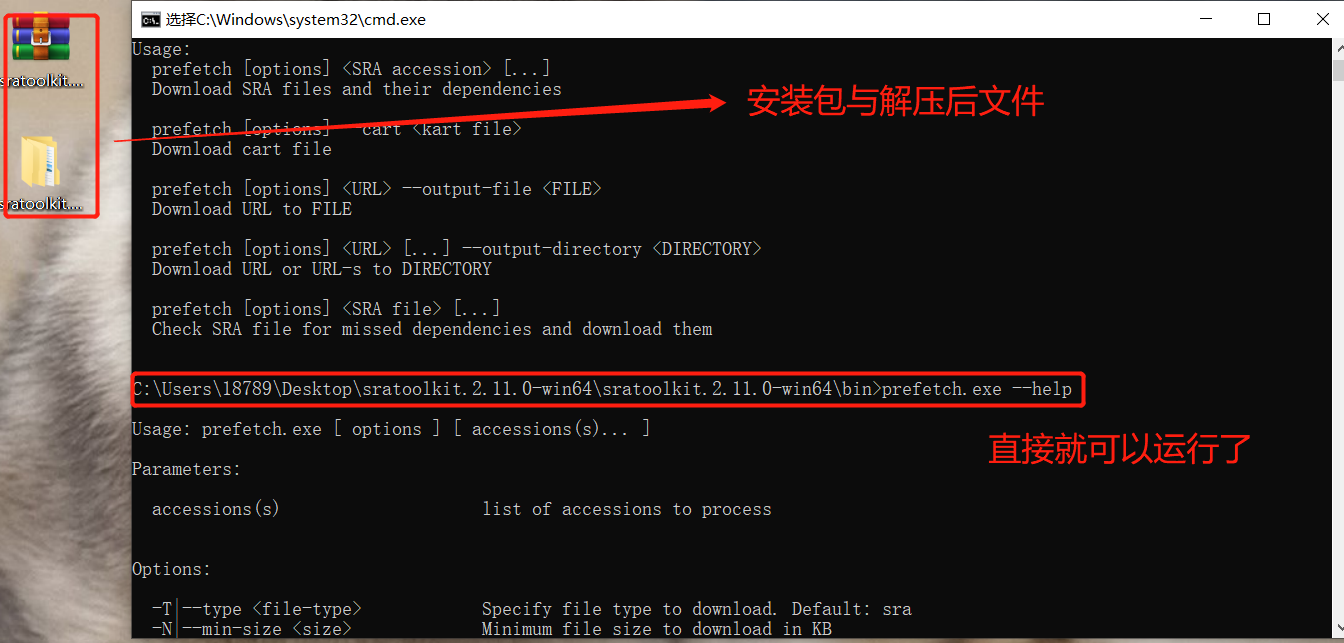
2. linux 下安装
1、下载:
直接下载安装包(在网页上下载),或者通过ftp协议下载(命令如下,比较慢)。
wget "http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz"
2. 解压
tar -zxvf sratoolkit.2.11.0-centos_linux64.tar.gz
3. 运行下载
以SRR2172038为例子,下载并转为fastq格式,和sam格式(常用工具)
#进入可执行程序目录下
cd sratoolkit.2.11.0-centos_linux64/bin
#下载 SRR2172038的数据,格式为sra格式. 下载完之后会出现SRR2172038.sra的数据
./prefetch SRR2172038
#利用 fasterq-dump 将sra格式转为fastq格式
./fasterq-dump SRR2172038.sra
#或者利用sam-dump转成sam格式
./sam-dump SRR2172038.sra
4. 批量下载
- 使用python脚本
# prefetch.py
import os
# SRR14067479-SRR14067488
for i in range(79,89):
id = 'SRR140674'+str(i)
print("正在下载SRR140674"+id)
os.system('prefetch -p '+id)
print("完成下载SRR140674"+id)
python prefetch.py # 直接运行
- 使用 SraAccList.txt
进入 all runs 详情页:
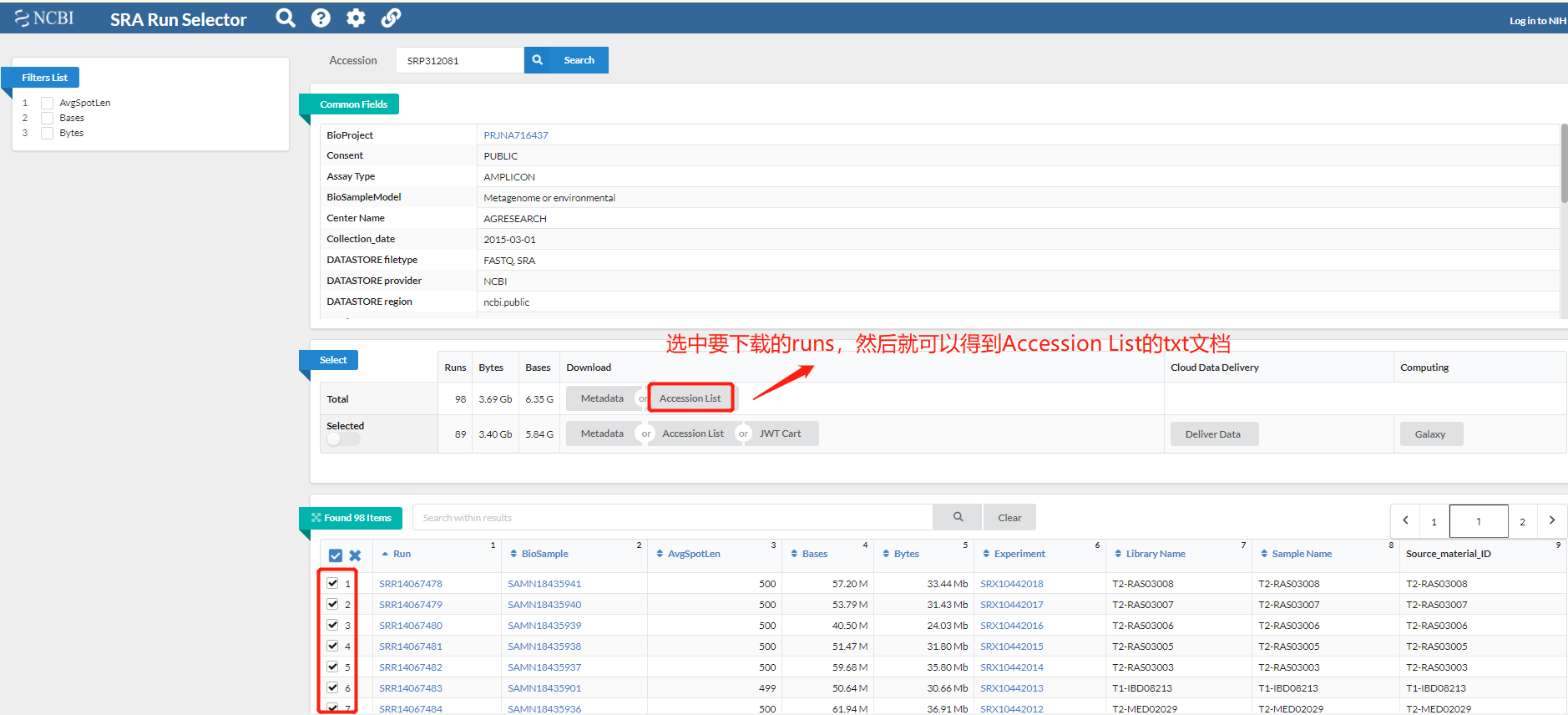
使用SRA TooKit 下载,(上面下载的txt文档为SraAccList.txt):
prefetch --option-file SraAccList.txt
备注:
-
这里只做简单演示。可以根据 模块的 --help参数,查看具体的参数进行使用。
-
可以将下载的SRA TooKit 加入到环境变量中,这样就可以在全局使用SRA TooKit
-
具体详情的信息,NCBI官网提供了相关的电子书籍
网址:https://www.ncbi.nlm.nih.gov/books/NBK56551/