
Quartet-puzzling算法与Likelihood Mapping分析
Quartet Puzzling算法
背景
在所有用于构建系统发育树的算法中,最大似然(ML)算法因其概念上的简单性以及明确的统计基础,而被广泛应用。
基本上,ML分析包含三部分:1. 确定一个核苷酸/氨基酸发生状态替代的模型;2. 基于该模型,通过计算某种进化历史的假设导致观测数据(比对序列)的概率,来评价各种进化历史的假设(包括进化树和树枝长度);3. 选择能够以最高概率产生观测数据的进化假说最为ML分析的结果,即确定最优树。
一个基因/蛋白从一条祖先序列开始,经过一段时间演变(发生位点状态替代),逐渐在子代序列中产生遗传多样性。这个演化过程,在概率统计上可以用平稳随机过程(马尔可夫过程,概称为进化模型)来描述。通过测序获得各种现存子代序列的序列组成,系统发育分析旨在重建自祖先序列到现存序列的演化过程。
关于演化过程的假说,可分为两部分:树拓扑结构(表达序列分歧关系)和树枝长度(表达序列分歧距离,等速率情形下还可表达分歧时间)。我们在选定一个进化模型时,实际上只是确定了一个分子进化的理论框架,例如,位点状态(或组分)频率是否一致?状态间替代速率是否一致?等。针对不同的序列,同一进化模型下,会有不同的模型参数。所以我们可以将进化模型理解为分子演化过程的数学表达形式,而我们要推定的进化树和各种枝长是分子演化的直观的图形表现。
所以在给定一条祖先序列和一个确定参数的进化模型时,在启动演化的“按钮”之后,这条祖先序列此后会变成什么样的子代序列是可预测的。那么,系统发育分析的过程恰好相反。利用一批对齐子代序列,反过来推定进化模型的相关参数,以及树拓扑和枝长。换句话说,就是用演化的结果来推测演化的过程。
前面我们说初始祖先序列和进化模型确定时候,演化的结果是可预测的。这背后隐含着一个概率论意义上的一个重要原理,即最大似然原理。进化模型与进化的结果依概率相关。
一个进化模型下,一个进化结果的出现实际上是一个概率事件。所以一个进化结果可以有不同的进化模式导致,只是不同的模式导致这一结果的概率不同。经验上我们会相信,目前结果的应该是导致该结果概率最高的进化模式产生的。这就是最大似然法的理论基础。
统计学上,最大似然法是参数估计中最重要的方法。在系统发育分析中,ML法也就是利用现有对齐数据,来对进化模型中的各种参数,以及树拓扑结构和枝长进行最大似然估计。
然而,进化模型中的较多参数,一棵树中的 $2N-3$个分枝,一组序列所有可能的树拓扑结构($\Pi_{i=3}^N (2i-5)$),这些都要进行ML估计,计算上的成本无疑是巨大的。所以在ML诞生之初,只能用于少量序列的进化树构建。此后,研究者开始考虑采取不同的策略来降低计算成本,其中包括以下两种思路:
- 用枝长的近似最大似然估计来代替真最大似然估计
- 用快速建树方法(如NJ)来代替树空间的搜索
这两种思路,特别是第二个一致沿用至今。1996年Strimmer和Haeseler提出了另外一种,降低ML算法计算成本的方法,即Quartet Puzzling算法。
QP算法
众所周知,构建进化树最小需要基于$4$条序列,而对于任何$4$序列,所有可能的二叉分枝的进化树拓扑结构仅有$3$种(下图中的$T_1,T_2,T_3$)。对于最少的$4$条序列,构建ML树时最为省时的。
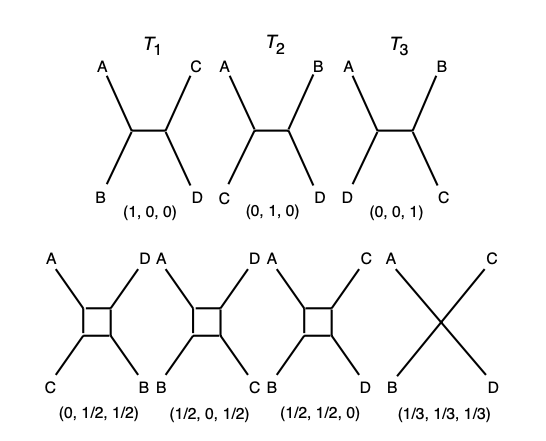
在一组$N$条序列构成的序列集合中,如果随机选取其中$4$条序列的话(称为一个 quartet),那么就有$C_n^4$中可能。因为ML分析的首要目标是确定一颗进化树,以及相关分枝的枝长。那么如果从任何一个含有$4$条序列的子集出发,构建其ML树,然后逐步往子树上添加剩下的序列。那么最终是后可能找到最优树的。
QP算法就是利用这样基本思想,来完成ML树的构建的。
Likelihood Mapping分析
原理
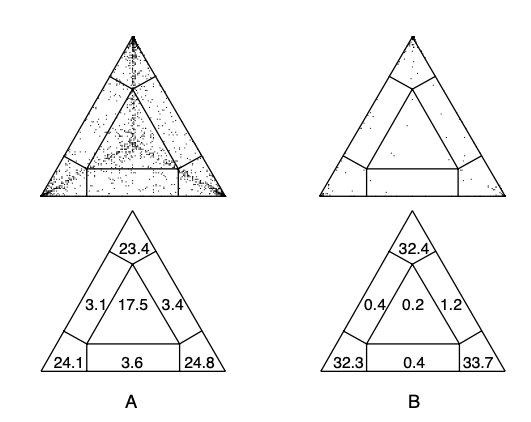
在利用QP算法构建ML树时,每个quartet都会有一个有三个概率值构成的向量$(p_1, p_2, p_3)$,其中的概率值,指的是这个quartet得到三种二叉分枝树的概率,所以其和等于1。如果$p_1$较大,这表明达到$T_1$的可能性较大。三种二叉树及其概率,可以直观的表达在一个如下图所示的三角图形上。
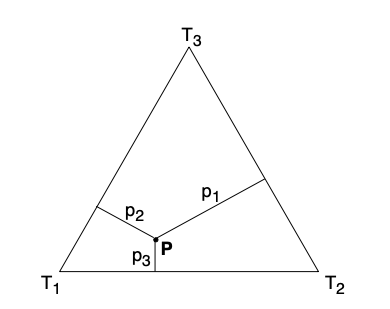
上图种的点$P$,到三条变的垂直距离分别表示了得到二叉树的概率。所以当某一个概率较大时,点$P$就会向三角形中代表对应二叉树的角落靠近。所以当三种二叉树的概率相等时,那么点$P$就会落在三角形的中心(下图A),这也就意味着该quartet的系统发育关系呈现星型结构,即不能解出序列之间的系统发育关系。进一步,我们可以将三角形的区域再细分(如下图B),点落在$A_1,A_2,A_3$区域内,表明该quartet的系统发育关系是相对明确的;点落在$A_{123}$内,表明该quartet系统发育关系是不明确的;而落在三个矩形区域内,则表明该quartet系统发育关系是部分明确的。
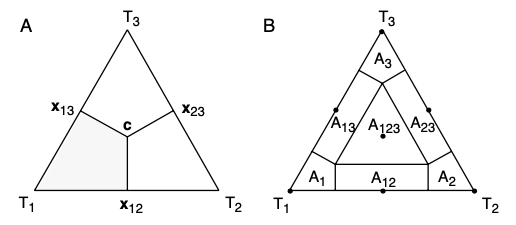
这就是Likelihood Mapping分析的基本原理。Likelihood Mapping是一种简单的、通过图形直观显示比对序列中有关系统发育信号的方法。将一组序列的所有quartet,通过在三角区域内打点的形式表达出来(如下图),可以直观的反映出该组序列数据的系统发育信号,例如关系可解的程度如何?等。
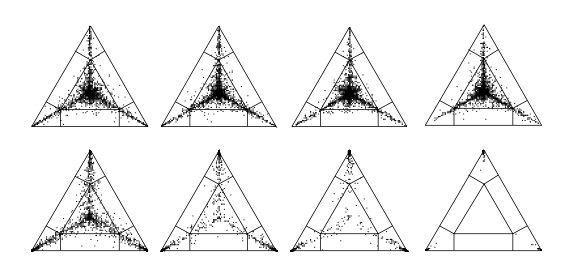
对于序列较多的数据集合,穷尽所有的quartet是没有必要的,随机选择其中1000个显出来,已经足以反映该组数据的系统发育结构信息了。
甚至,我们可以用分布各区域内的点的比例,还进行定量的描述,如下图所示。
Four-cluster likelihood mapping
上述likelihood mapping方法有一个更常见的应用场景,即Four-cluster likelihood mapping。在一组序列中,我们往往关心的其系统发育树中,某些子类群(分枝之间)之间的系统发育关系。因此,按照likelihood mapping的思路,我们可以将quartet的选择,限定在4个序列群(sequence cluster)内,而这4个序列群分别对应一个子类群(子分支)。
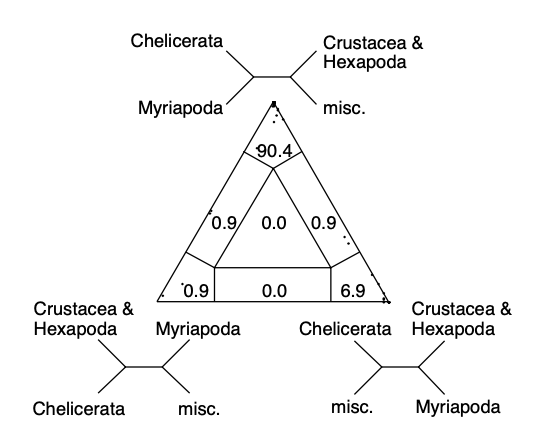
在上图所示的例子中,有90.4%的quartet支持顶部的拓扑结构的。
Tree-Puzzle
QS算法构建进化树,和likelihood mapping需要借助软件Tree-Puzzle。
Tree-Puzzle需要下载源码编译安装。下面我们用Tree-Puzzle的并行版本来演示其用法。
(base) [user@server data]# mpirun -np 10 ppuzzle ## 启动ppuzzle后,软件提示输入 比对序列文件
WELCOME TO TREE-PUZZLE 5.3.rc16!
Using SPRNG -- Scalable Parallel Random Number Generator
RANDOM SEED: 439925973
Please enter a file name for the sequence data: EF.phy
Input data set (EF.phy) contains 22 sequences of length 915
1. EFTU_ECOLI
2. EFTU_HELPY
3. EFTU_MYCGE
4. EF1A_SULAC
5. EF1A_DESMO
6. EF1A_AERPE
7. EF11_HUMAN
8. EF12_MOUSE
9. EF1A_CAEEL
10. EF11_DAUCA
11. EF1A_DICDI
12. EF2_SULAC
13. EF2_AERPE
14. EF2_DESMO
15. EF2_MOUSE
16. EF2_HUMAN
17. EF2_CAEEL
18. EF2_DICDI
19. EF2_BETVU
20. EFG_ECOLI
21. EFG_HELPY
22. EFG_MYCGE
(consists very likely of amino acids encoded on nuclear DNA)
GENERAL OPTIONS
b Type of analysis? Tree reconstruction ## 选择分析类型,默认为构建进化树,输入b回车后,将设定为Likelihood mapping
k Tree search procedure? Quartet puzzling
v Quartet evaluation criterion? Approximate maximum likelihood (ML)
n Number of puzzling steps? 1000
o Display as outgroup? EFTU_ECOLI (1)
z Compute clocklike branch lengths? No
e Parameter estimates? Approximate (faster)
x Parameter estimation uses? Neighbor-joining tree
OUTPUT OPTIONS
9 List puzzling trees/splits (NEXUS)? No
u List unresolved quartets? No
j List puzzling step trees? No
SUBSTITUTION PROCESS
d Type of sequence input data? Auto: Amino acids
m Model of substitution? Auto: LG (Le-Gascuel 2008)
f Amino acid frequencies? Estimate from data set
RATE HETEROGENEITY
w Model of rate heterogeneity? Uniform rate
Quit [q], confirm [y], or change [menu] settings: b ## 输入b,选择分析类型
GENERAL OPTIONS
b Type of analysis? Likelihood mapping
v Quartet evaluation criterion? Exact maximum likelihood (ML)
g Group sequences in clusters? No ### 是否对序列分组,即是否使用Four-cluster likelihood mapping
n Number of quartets? 5 (all possible)
e Parameter estimates? Approximate (faster)
x Parameter estimation uses? Neighbor-joining tree
SUBSTITUTION PROCESS ## 模型设置
d Type of sequence input data? Auto: Binary states
m Model of substitution? Two-state model (Felsenstein 1981)
f Binary state frequencies? Estimate from data set
RATE HETEROGENEITY ## 模型设置
w Model of rate heterogeneity? Uniform rate
Quit [q], confirm [y], or change [menu] settings: g ## 开启序列分组
Number of clusters (2-4): 4 ## 将序列分为4组
Distribute all sequences over the four [c]lusters [a], [b], [c], and [d] ## 每组的标记分别为 a, b, c, d
(at least one sequence per cluster is necessary),
type [x] to exclude a sequence and [q] to get back to the menu:
EFTU_ECOLI: a # 将序列ID为Gibbon的序列放入 a 组
EFTU_HELPY: a # 依此类推
EFTU_HELPY: a
EFTU_MYCGE: a
EF1A_SULAC: b
EF1A_DESMO: b
EF1A_AERPE: b
EF11_HUMAN: b
EF12_MOUSE: b
EF1A_CAEEL: b
EF11_DAUCA: b
EF1A_DICDI: b
EF2_SULAC : c
EF2_AERPE : c
EF2_DESMO : c
EF2_MOUSE : c
EF2_HUMAN : c
EF2_CAEEL : c
EF2_DICDI : c
EF2_BETVU : c
EFG_ECOLI : d
EFG_HELPY : d
EFG_MYCGE : d
Number of sequences in each cluster:
Cluster a: 3
Cluster b: 8
Cluster c: 8
Cluster d: 3
Excluded sequences: 0
GENERAL OPTIONS
b Type of analysis? Likelihood mapping
v Quartet evaluation criterion? Approximate maximum likelihood (ML)
g Group sequences in clusters? Yes (4 clusters as specified)
n Number of quartets? 576 (all possible)
e Parameter estimates? Approximate (faster)
x Parameter estimation uses? Neighbor-joining tree
SUBSTITUTION PROCESS
d Type of sequence input data? Auto: Amino acids
m Model of substitution? Auto: LG (Le-Gascuel 2008)
f Amino acid frequencies? Estimate from data set
RATE HETEROGENEITY
w Model of rate heterogeneity? Uniform rate
Quit [q], confirm [y], or change [menu] settings: y ## 分组完成后,输入 y确认所有选项,并开始运行ppuzzle
Writing parameters to file EF.phy.puzzle
Writing parameters to file EF.phy.puzzle
Writing pairwise distances to file EF.phy.dist
Writing parameters to file EF.phy.puzzle
Writing parameters to file EF.phy.puzzle
PPP1: 0 (./puzzle1.c:8690)
Performing likelihood mapping analysis
PPP1: 1 (./puzzle1.c:8696)
All results written to disk:
Puzzle report file: EF.phy.puzzle
Likelihood distances: EF.phy.dist
Likelihood mapping diagram (EPS): EF.phy.eps # Likelihood mapping的结果
Likelihood mapping diagram (SVG): EF.phy.svg # Likelihood mapping的结果
The parameter estimation took 0.00 seconds (= 0.00 minutes = 0.00 hours)
The computation took 2.00 seconds (= 0.03 minutes = 0.00 hours)
including input 148.00 seconds (= 2.47 minutes = 0.04 hours)
参考文献
Strimmer, K., and A. von Haeseler (1996) Quartet puzzling: A quartet maximum likelihood method for reconstructing tree topologies. Mol. Biol. Evol. 13: 964-969. DOI: 10.1093/oxfordjournals.molbev.a025664
Korbinian Strimmer und Arndt von Haeseler. 1997. Likelihood-mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA 94:6815–6819. DOI: 10.1073/pnas.94.13.6815
Schmidt, H.A., K. Strimmer, M. Vingron, and A. von Haeseler (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 18:502-504. DOI: 10.1093/bioinformatics/18.3.502