
利用P4构建Bayes树
关于 P4
P4是用于分子序列最大似然和贝叶斯分析的Python软件包。 它的特点是可以使用异构模型。
P4也是系统发育工具包,接口是Python编程语言,因此P4具有高级面向对象编程语言的所有能力。 这是一件好事,但这意味着您需要了解一些Python才能使用它。 它可以对树或数据进行编程操作。
源码下载地址:https://github.com/pgfoster/p4-phylogenetics
P4的用法有交互式和批处理式(编写脚本程序)两种。
从read()开始
将系统发育分析相关数据和树读取到P4中的方法是用read()函数。不管在脚本程序中还是在P4交互环境中,read()函数都不返回任何东西。当我们读取一个序列比对文件时,一个Alignment对象将被生成,并保存在var.alignments中。var.alignments是一个列表,所以P4的一般打开方式是这样的:
#脚本模式
from p4 import *
read('myAlignment.phylip')
a = var.alignments[0]
read('myTreeFile.nwk')
t = var.trees[0]
序列比对与数据
通常情况下,我们进行系统发育分析,都仅需要一个序列比对文件。如上所示当通过read()函数读取序列比对文件后,其内容会保存在列表var.alignments中。而如果read()函数读取的序列不等长,那么内容则会保存在列表var.sequenceLists中。
在P4中有三个不同的序列数据相关的类:
- Alignment, 是SequenceList的子类, 有很多函数方法;
- Part, 数据分区,用于似然计算。 每个Alignment至少有一个Part,或更多。
- Data, 要使用的所有分区。它至少是一个Alignment (但可能更多),至少有一个Part (每个Alignment至少有一个Part,或更多)。 似然计算需要一个Data对象。 它具有序列的C语言表示,作为分区的列表。
一个含有系统发育数据的比对文件被读取为一个Alignment对象,这将非常有利于一些针对Alignment的简单任务。但是,如果你想计算数据的似然值,那么用Alignment对象来做就会很复杂,我们需要将Alignment转变为C语言形式。Part对象封装了C形式的Alignment(或Alignment分区);而Data对象则包含要使用的所有Part对象,以及这些Part所来自的Aignment。
在通过read()函数读取比对序列文件后,为了便于似然计算,可通过Data()函数还获得Data对象。
from p4 import *
read('myAlignment.phylip')
d = Data()
如果你想要使用的数据在多个比对序列文件中,这对于P4是完全可以接受的。
read('myFirstAlignment.phy')
read('mySecondAlignment.phy')
d = Data()
模型描述
所有的分子进化模型,总的来说都包含以下四部分内容:
- 一个表示各组分(核酸,氨基酸)频率的向量。
- 一个表示组分替代速率的矩阵。
- 不变位点的比例。
- 描述位点间替代速率差异的离散Gamma分布
在P4中设定进化模型,则是通过以上四点分别设置相关参数的。下面是F81模型(无位点间替代速率差异)的设置:
read('myAlignment.phy') # 读取序列比对文件
d = Data() # 获得Data对象
read('myTreeFile.nwk') # 读取一个树文件
t = var.trees[0]
t.data = d # 将比对的Data对象附加到tree的data属性上。
# 下面开始设置模型
t.newComp(free=1, spec='empirical')
t.newRMatrix(free=0, spec='ones')
t.setNGammaCat(nGammaCat=1) # no 'gamma'
t.setPInvar(free=0, val=0.0) # no proportion of invariant sites
t.calcLogLike() # 计算在以上所设模型下myTreeFile.nwk对myAlignment.phy的似然值。
模型的参数可能是自由地从实际数据中优化free=1;也可以不这样操作free=0,但需要通过spec来指定。当spec='specified'时,还须通过val来自定义参数的具体值。
t.newComp(free=0, spec='specified', val=[0.4, 0.3, 0.2]) # 四组分频率,分别为0.4,0.3,0.2,0.1
对组分频率的设定(tree.newComp(partNum=0, free=0, spec='empirical', val=None, symbol=None)),spec可设定为:
- 'empirical' no val, free=1
- 'equal' no val, free=0
- 'specified' val=[aList], free=0
- 'wag' no val, free = 0 (cpREV, d78, jtt, mtREV24, mtmam, wag, etc)
对替代速率矩阵的设定,通过tree.newRMatrix(partNum=0, free=0, spec='ones', val=None, symbol=None)完成。其中spec可选项有:
- ‘ones’ - for JC, poisson, F81
- ‘2p’ - for k2p and hky
- ‘specified’
- ‘cpREV’
- ‘d78’
- ‘jtt’
- ‘mtREV24’
- ‘mtmam’
- ‘wag’
- ‘rtRev’
- ‘tmjtt94’
- ‘tmlg99’
- ‘lg’
- ‘blosum62’
- ‘hivb’
- ‘mtart’
- ‘mtzoa’
当spec=specified或spec=2p时,val须被指定;其中当spec=2p时,val=kappa;当spec=specified时,val须通过一个列表将所组分替代速率,列表长度为(((dim * dim) - dim) / 2) - 1,对DNA而言,dim=4,所以列表中有5个元素。
设定参数最复杂的GTR模型时,须将组分频率和替代速率都设定为自由优化,即
t.newComp(free=1, spec='empirical')
t.newRMatrix(free=1, spec='ones')
对不变位点比例的设定,则通过tree.setPInvar(partNum=0, free=0, val=0.0)完成。free=0时,意味着模型中无+I项,即无不变位点比例的设定。相反free=1时程序将通过数据来优化+I参数。
对模拟位点间替代速率差异的离散Gamma分布的参数设定,由tree.setNGammaCat(partNum=0, nGammaCat=1)和tree.newGdasrv(partNum=0, free=0, val=None, symbol=None)两个函数完成。
t.setNGammaCat(nGammaCat=4)
t.newGdasrv(free=1, val=0.5)
无以上两项设定,或设定为默认参数,则意味着模型中无+G4项。
Tree-heterogeneous models
以上关于分子进化模型的设定,都属于同构模型,这在其他很多软件中都能实现,并无特别之处。然而之所以学习P4的建树方法,是因为它能使用异构模型。
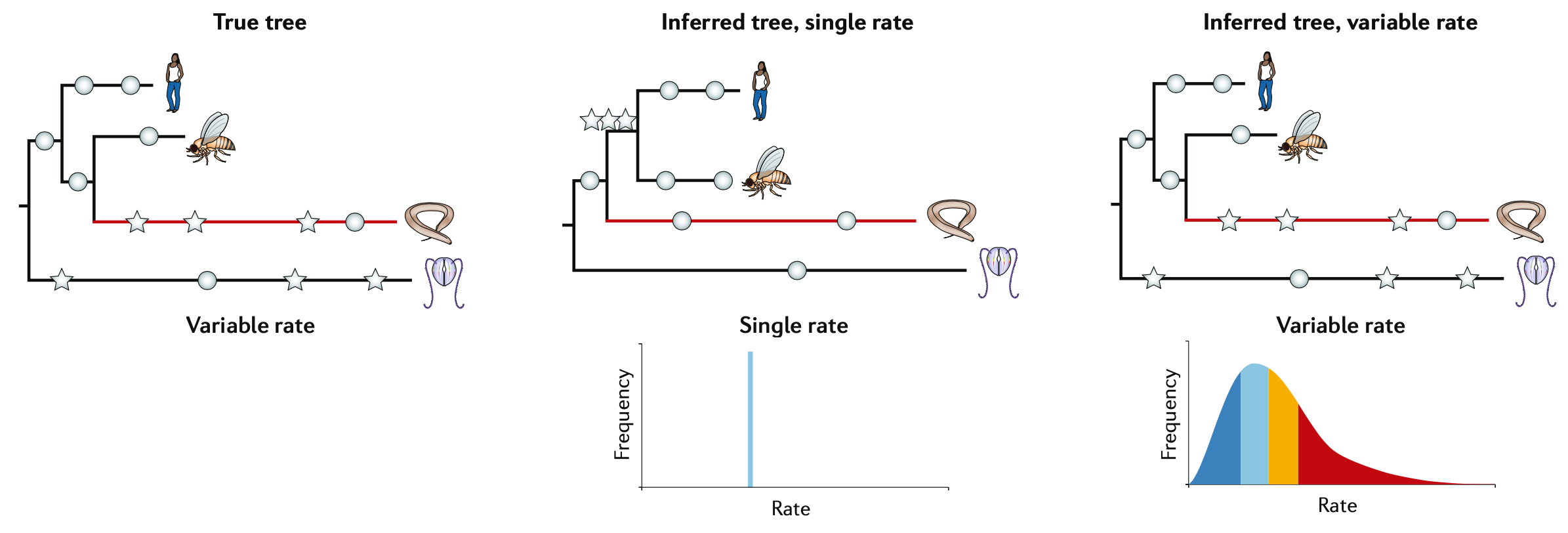
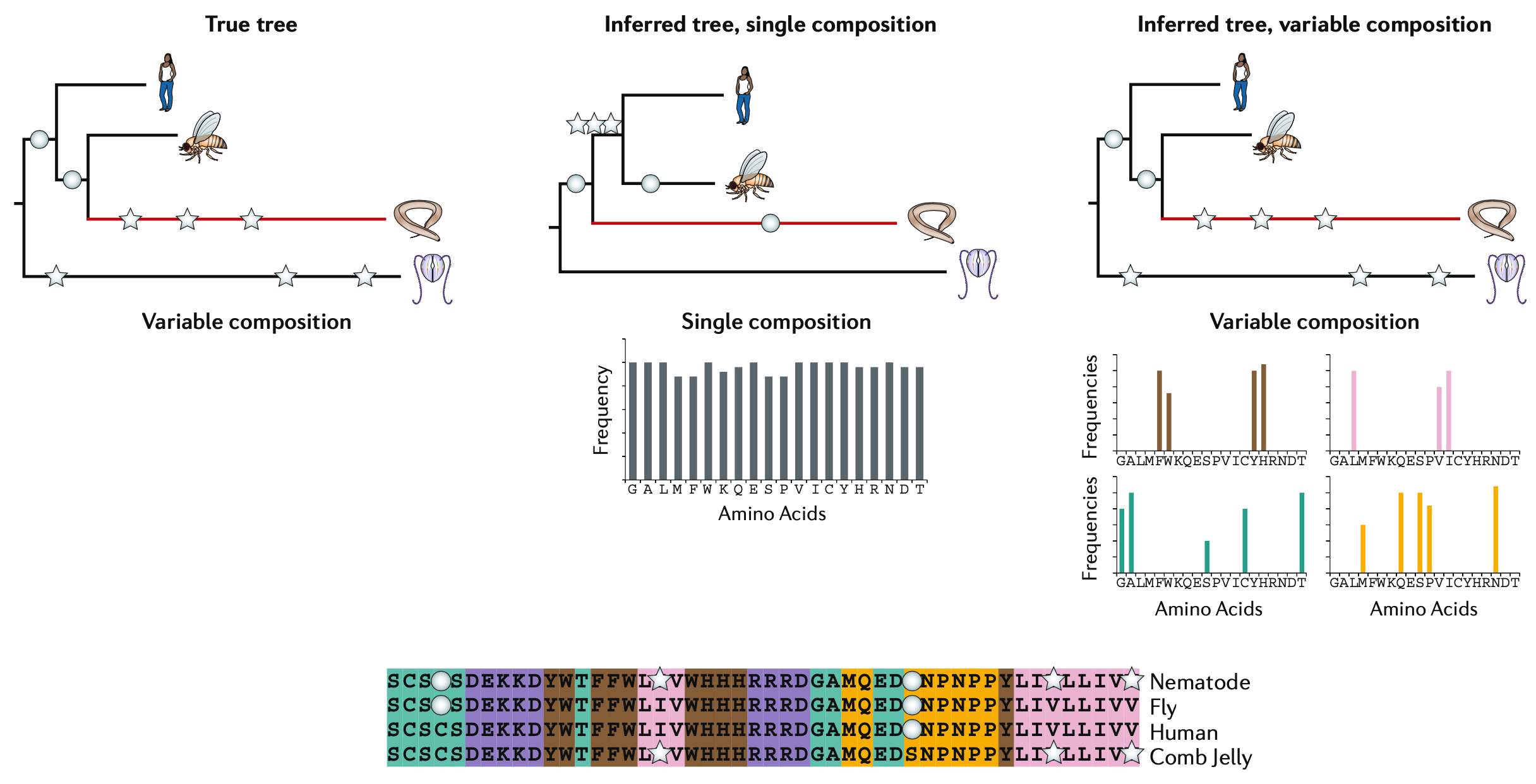
图引自 DOI: 10.1038/s41576-020-0233-0
所谓异构模型,或树内异构模型,即:在由序列代表的物种所有构成物种树内,不同的分枝上允许有多个不同的组分频率的向量和/或多个不同的组分替代速率矩阵。而同构模型则只有一个组分频率的向量和一个组分替代速率矩阵。
假设现有4个物种的进化树,我们要为clade 1设定一个组分频率向量,而其他分枝都用另一个组分频率向量,可通过以下方式实现。
p4> read("((one,two),(three,four));") # 非文件名的读取,P4会有警告信息,可通过var.warnReadNoFile=False关闭。
p4> t = var.trees[0]
p4> t.draw()
+--------2:one
+--------1
| +--------3:two
0
| +--------5:three
+--------4
+--------6:four
p4> A = t.newComp(free=1, spec='empirical', symbol='A') # 注意!这里只是演示方法,在实际操作中设定模型前,需要首先为t.data附加Data对象
p4> B = t.newComp(free=1, spec='empirical', symbol='B')
p4> t.setModelThing(A, node=t.root, clade=1) # 将组分频率向量A,从树根节点开始,赋予所有子分枝(如clade=0,则只针对与根节点直接相连的分枝)
p4> t.setModelThing(B, node=1, clade=1) # 将组分频率向量B,从节点1开始,赋予所有子分枝
p4> t.draw(model=1)
+BBBBBBBB2:one
+BBBBBBBB1
| +BBBBBBBB3:two
0
| +AAAAAAAA5:three
+AAAAAAAA4
+AAAAAAAA6:four
以上是比较明确的为某些分枝指定不同的模型的方法,同理我们还可以为不同的分枝设定不同替代速率矩阵。
此外,P4还提供了另一种半随机的方式。
p4> t.newComp(free=1, spec='empirical')
p4> t.newComp(free=1, spec='empirical')
p4> t.setModelThingsRandomly()
Metropolis-coupled MCMC
通过读取数据,设定相关进化模型后,即可使用Metropolis-coupled MCMC算法,进行Bayes分析,确定进化树相关参数,构建进化树。P4执行MCMC的方式如下:
p4> # t is the tree, with model and data attached
p4> m = Mcmc(t, nChains=4, runNum=0, sampleInterval=1000, checkPointInterval=250000, simulate=2)
p4> m.run(1000000) # one million generations
nChains=4表明P4的MCMCMC将同时启动4条链,其中一条为冷链,三条热链,这在MrBayes软件中有较为详细的介绍。在MCMCMC运行过程中,所有的树,似然值,以及模型参数都会分别保存在不同的文件中,他们都将用于此后对MCMC结果的评价。下面脚本是一个简单但完整的例子:
read("myalignment.phy") #读取序列比对文件
d = Data() # 提取Data对象
t = func.randomTree(taxNames=d.taxNames) # 基于Data对象生成一个随机树
t.data = d # 将Data对象附加于随机树
# 设定模型,GTR+I+G4+F
t.newComp(free=1, spec='empirical')
t.newRMatrix(free=1, spec='ones')
t.setNGammaCat(nGammaCat=4)
t.newGdasrv(free=1, val=0.5)
t.setPInvar(free=1, val=0.2)
# 4条链,运行1000000代,每500代取样一次,将共取2000个样本,每个250000代存一个checkpoint
# 如果在同一目录下想要运行多个mcmc,那么不同的mcmc需要设定不同的runNum=0,1,2,...
m = Mcmc(t, nChains=4, runNum=0, sampleInterval=500, checkPointInterval=250000)
# Here you can adjust the proposal probabilities, and tuning parameters.
m.prob.polytomy = 1.0 # Default is zero
m.prob.brLen = 0.001 # Default is zero
m.autoTune() #开启参数的自动微调
# 开始运行mcmc
m.run(1000000)
关于checkpoint
checkPointInterval 需要能被总运行代数整除
在MCMC运行过程中,按照函数参数的的设定,每个250000代会保存一个checkpoint。保存checkpoint的好处,是可以在程序中断时,可以接着上一个checkpoint的继续(通过func.unPickleMcmc()函数),又或者可以在完成一次MCMC后,继续延长该MCMC。
因为每一个checkpoint是一个完整的Mcmc对象,因此没有Data对象,所有在从checkpoint重启mcmc时,需要给func.unPickleMcmc(0, d)函数传递一个Data对象,如果是重新进入P4的,那么就需要重新读取文件,如:
p4> read("myalignment.phy")
p4> d = Data()
p4> m = func.unPickleMcmc(0, d) # runNum 0
# 在运行之前,还可对m (Mcmc对象) 进行参数调整
p4> m.run(1000000) # 接着上一个保存的checkpoint,再运行1000000代。
如果mcmc意外终止,那么在接着上一个保存的checkpoint继续运行前,需要将相关文件中上一个checkpoint之后产生的数据删除!
关于mcmc的评价
对于mcmc结果的评价,有时我们会通过看对数似然值的走势是否进入稳定的平台期,来判断mcmc已经收敛。但该方法并不始终稳定可靠。因为虽然未进入平台期即为收敛,但反之不尽然。因此,P4采用了和PhyloBayes类似的做法,即比较不同的checkpoint,或不同的run之间的split supports来评价mcmc。
从一次mcmc run中读取所有checkpoint,需要通过函数McmcCheckPointReader(fName=None, theGlob='*', last=False, verbose=True),
p4> cpr = McmcCheckPointReader()
p4> m = cpr.mm[0] # 得到第一个checkpoint
p4> cpr.compareSplits(0,1) # 比较第一个和第二个checkpoint间的split supports
p4> cpr.compareSplitsAll() # 比较所有checkpoint间的split supports
p4> cpr.writeProposalAcceptances() # 输出proposal acceptances
根据MrBayes作者的建议 proposal acceptances 应该在10-70%,是比较理想的结果。
从cpr.compareSplitsAll()可以看到两两average standard deviation of split support (ASDOSS)值,该ASDOSS < 0.01则认为该mcmc已经收敛。
通常情况下,我们都会运行多个mcmc run。那么评价多个mcmc run,首先需要读取多个run的数据。如前所示,在当前目录下只有一个run的数据是,执行McmcCheckPointReader()则只读取一个run的数据。而如果当前目录中有多个run的数据,McmcCheckPointReader()则会读取所有run的数据,而此时入要单独读取某一个run的数据,则需要通过fName=0或theGlob='*_0.*'来指定。
读取多个run的数据之后,即可用compareSplitsAll()评价各mcmc run。
输出最终结果
经过这一番操作,我们最终的目的是得到一颗Bayes进化树。在运行mcmc,每个500代,程序会进行一次采样,包括树、似然值和模型参数,并分别保存在不同的文件中。假设分别独立运行了两次mcmc,那么当前目录中就会有两个有关树的文件:mcmc_trees_0.nex和mcmc_trees_0.nex。
首先我们可以通过TreePartitions()函数将这些树读到P4环境中来,在通过TreePartitions对象的read()函数将第二个run的树合并进来。
p4> tp = TreePartitions('mcmc_trees_0.nex', skip=1000) # 跳过前1000个树 burnin
p4> tp.read('mcmc_trees_1.nex', skip=1000) # 跳过前1000个树 burnin
p4> t = tp.consensus() # 获得一致树
p4> for n in t.iterInternalsNoRoot(): # 将分枝支持度转为百分制
n.name = '%.0f' % (100. * n.br.support)
p4> t.writeNewick('bayes_cons.nwk') # 保存树
参考资料
http://p4.nhm.ac.uk/index.html
Foster P. 2004. Modeling compositional heterogeneity. Systematic Biology 53: 485–495. DOI:10.1080/10635150490445779