
生物信息学简史
生物信息学
众所周知,现代生物信息学是利用计算机分析软件来分析生物分子数据的一门科学。现如今,信息大爆炸的时代,计算机已然渗透到每个自然科学中,利用计算机辅助完成一些基础研究工作早已习以为常。唯独生物学用“生物信息学”这个特定的名词来定义生物学研究中利用计算机科学辅助分析。究其缘由,有两点,其一是生物学本身是被认为是“硬科学”(通常指自然科学)与“软科学“(通常指社会科学)的交界面。其二是与生物信息学发展历程相关的。
生物信息学发展历程
起源:蛋白质序列分析
现在我们熟知的生物信息学是利用桌面计算机辅助完成生命科学研究,但其起源却要追溯到没有桌面计算机的20世纪60年代。
1958年到1960年,世界上第一个广为人知的生物信息学软件--COMPROTEIN诞生了。COMPROTEIN,由生物信息学的奠基人Margaret Dayhoff和她当时的同事Robert S. Ledley开发,用于拼接当时盛行的Edman降解法测蛋白质序列所产生的大量短的肽链序列。1965年,Dayhoff和Eck建立了第一个只包含65条蛋白质序列的生物序列数据库--Atlas of Protein Sequence and Structure。


1963年,Emile Zuckerkandl 和 Linus Pauling提出了关于可以利用直系同源蛋白序列之间的变化来重建其进化历史的概念。从而引出了一个生物信息学新的方向,序列比对。但当时没有具体评价蛋白质序列进化价值的比对算法。同时由于20世纪70年代生物信息学的重心偏向于DNA序列分析发展,导致氨基酸的替代模型直到1986年才问世。1986年,Dayhoff、Schwartz和Orcutt历时八年,建立了第一个氨基酸替代模型--PAM1。该模型基于在71个同源性高于85%的蛋白质家族的系统发育树中观察到1572个点接受突变(PAM)。得到的结果是20×20不对称替代矩阵,该矩阵包含基于观察到的每个氨基酸突变的概率值(每个氨基酸在给定的进化间隔内变化的概率)。

转变:DNA序列分析
随着Francis Crick的中心法则的提出,DNA成为生命科学中最重要的生物大分子。自然而然的,人们对于生命科学研究的重心逐渐转变为DNA。
1977年,Sanger和他的团队发明了DNA测序技术并随后改进为如今的第一代测序技术--Sanger化学终止法。
测序技术的诞生,使得DNA的碱基组成不再是秘密。但是,DNA的碱基组成中蕴含着怎样的遗传信息?这些遗传信息之间的差异如何反映在进化中?我们还不得而知。所以,对于DNA序列的中遗传信息的提取也就是DNA序列分析变得尤为重要。DNA分析的一般包括:
- 序列比对(比如,找到不同物种之间序列的同源序列)
- 计算(比如,利用PAM1替代矩阵来重建多直系同源蛋白的系统发育树)
- 模式匹配(比如,DNA编码序列的开放阅读框的识别)
1979年,Roger Staden开发了第一个生物信息学软件用于分析Sanger序列。Roger的Staden Package主要用来Sanger序列的拼接和注释。并且其至今仍在更新和使用。
DNA上包含编码与非编码序列,而编码序列往往连续的分布在少数的DNA分子上。所以基因无法实现生化分离,而且丰富度远低于其编码产品。同时给基因分析带来了一定阻碍。
1972年,Jackson, Symons 和 Berg利用限制性核酸内切酶和DNA连接酶剪切环状的SV40病毒DNA并插入到lambda DNA,随后转染到大肠杆菌中,实现了基因的分离和扩增。1985年,Kary Millus发明了PCR,使基因的扩增变得高效和便捷。同时,PCR如今也广泛运用于DNA文库的构建中。
计算机科学的发展
随着DNA测序技术和基因分子操作技术的诞生,DNA序列信息的挖掘成为生命科学研究的主流。但是面对庞大数量的DNA序列数据,人类本身几乎无法数据处理。而计算机自诞生以来就是辅助人类完成一系列复杂和重复性的工作。所以计算机科学的发展也影响着生物学的发展。
桌面计算的发展
桌面计算机的发展主要是两方面,一个是体积的缩小,另一个是操作系统的人性化(操作的简便化)。
1965年,体积相当于冰箱重达180磅的DEC PDP-8微型计算机。

1974年,世界上第一台桌面计算机的Intel 8080微型处理器。这些计算机都很难面向大众。1977年,Commodore PET,、Apple II 和 Tandy TRS-80三台微型计算机以价格低,体积小,操作简便赢得了市场,成为真正面向大众的计算机。



20世纪80年代到如今,一系列的个人计算机和小型工作站相继问世。比如1984年DEC VAX-11,HP-9000,1985年的CP/M和 Macintosh等。 计算机逐渐普及化,为生物信息学的发展奠定基础。


专业软件的发展
计算机的体积的缩小和价格的降低,使得科学中应用计算机的成本大幅降低。但对于生命科学来说,专业软件的发展也是必不可少的。
从1958年Dayhoff首次在生命科学研究中应用计算机设计出第一个生物信息学软件开始,不断有新的分析软件的诞生。
| Year | 1958 | 1979 | 1984 | 1985 | ... |
|---|---|---|---|---|---|
| Name | COMPROTEIN | Staden Package | GCG | DANSTAR | ... |
| Description | The first bioinformatic software | The first software to analysis Sanger sequences | Manipulating DNA, RNA or protein sequences. | Running on a personal computer | ... |
编程语言的发展
专业软件的发展离不开编程语言的发展。从桌面计算机和工作站开发到普及,编程语言也随之在不断改善。
20世纪80年代中期,生物信息学领域流行使用一些脚本语言,这些语言至今仍在使用。但是脚本程序(无需编译)往往执行起来比较费时间。
1994年,由Larry Wall 1987年开发的Perl语言,出现在生物信息学领域,并开始流行起来。
2000年,Python逐渐走进生物信息学家的视野,由于简单的语法,Pytho逐步取代Perl成为生物信息学的主流编程语言。
免费化
如今,几乎所有的生物信息学分析软件都是开源的,免费提供下载。这极大的方便了生物信息学研究,为生物信息学科的发展奠定基础。
提倡免费化最早是一个计算机科学的运动--GNU宣言。1985年,Richard Stallman发表了GNU宣言。宣言倡导提供免费的基于UNIX的操作系统。这次免费化的运动推动了免费软件基金会的成立。
网络时代的来临
科学的研究离不开合作,一个团队的合作,一个国家的合作甚至整个世界的合作。合作中最重要的问题便是信息交流。20世纪90年代初,Tim Berners-Lee开发了名为万维网(World Wide Web)的全球信息系统。万维网的建立使得科学的交流不在收地区的限制。生物信息学的许多软件都是在线上提供下载等。
超级计算机的诞生
尽管在大多数情况下,一台简单的台式计算机就可以满足计算的要求。但随着结构生物学的诞生,分析序列信息的计算级别显然满足不了其对于生物大分子的动态模拟。所以,高性能计算的超级计算机的应用解决了这一需求。
- Compute Canada
- New York State’s High Performance Computing Program
- The European Technology Platform for High Performance Computing
- China’s National Center for High-Performance Computing.
高性能的计算的需求同样使得像Amazon和Microsoft这样的公司提供生物信息学服务。
组学时代
1991年,美国国家卫生研究院(National Institutes of Health)发起了标志着走入基因组时代的人类基因组计划(Human Genome Project,HGP)。HGP,前后包括英、美、法、中等六个国家参与,历时13年花费27亿美金才最终完成。

随着基因组时代的到来,许多全基因组序列拼接生物信息学软件相继问世。
高通量生物信息学
二代测序技术的诞生以来,序列数据的数量几乎是指数级的扩增。
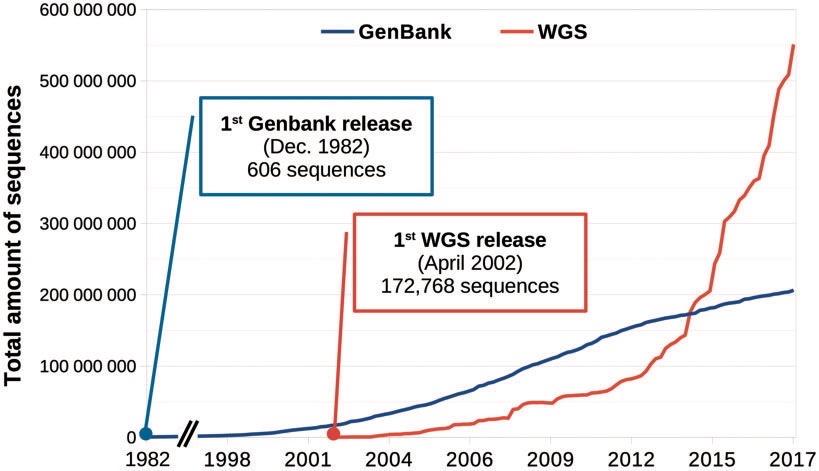
如今,我们熟知的大大小小的数据库中保存着亿万级别的数据信息。大数据时代背景下,序列信息的提取分析成为主流。
二代测序技术
1991年,测序人类基因组(3.3Gb)需要花费13年,27亿美金才能完成。然而2018年,测序人类基因组只需1000美金,一周的时间。这便是二代测序技术的诞生所带来的改变。
最初的二代测序技术便是454焦磷酸测序技术。454焦磷酸测序技术可以同时测成千上万的DNA序列,极大的提高了测序的效率。
如今,市面上已经存在多种高通量的测序技术。随之而来的便是多种处理特有的测序技术的生物信息学工具。
生物信息学大数据
作为信息时代的今天,大数据这一词对所有人都不陌生。许多行业,领域都会采集并分析属于他们的大数据。那么生物信息学作为与计算机科学联系紧密的学科自然也不例外。
由于高通量的测序技术的出现和发展、网络系统的建立,生物信息学也进入大数据时代。处理分析基因组、单个生命体的全基因组、单个物种的泛基因组甚至特定环境的宏基因组等这样的组学序列数据或生物进化相关的基因家族序列数据是如今生物信息学的主题。
数据库的开放和共享
人类目前来说无法去创造自然界本不存在的生命形式,只能通过不断的去发现自然界中生命的组成来探寻生命的奥秘。作为利用序列信息去窥探生命的生物信息学,存储这些生物大分子的序列信息便是发展的基础。
1968年,Dayhoff和Eck建立了第一个只包含65条蛋白质序列的生物序列数据库--Atlas of Protein Sequence and Structure。Dayhoff随后便是利用这一数据库的蛋白质序列信息开发了氨基酸替代模型PAM。
数据库的开放和共享也为生物信息学的发展推波助然。1986-1987年,欧洲分子生物学库(EMBL)、Genbank和日本DNA数据银行(DDBJ)序列数据库,为了能够标准化数据格式、定义发表的核苷酸序列最小信息和方便数据库之间数据的分享先后联合。如今,这个联盟被称为国际核苷酸序列数据库集(International Nucleotide Sequence Database Collaboration,INSDC)。
随着网络时代的到来,一些序列数据库纷纷上线。
| Database | Genbank | EMBL | NCBI(Including BLAST) | Genomes | PubMed | Human Genome | ... |
|---|---|---|---|---|---|---|---|
| Online Year | 1992 | 1993 | 1994 | 1995 | 1997 | 1999 | ... |
未来的展望:迈向整个生物体建模时代-系统生物学
伴随着各种分子生物学的技术和计算机科学的并行发展,生物信息学从最初的蛋白质序列拼接、单独的DNA序列分析到组学序列的分析、大数据时代下的序列分析再到如今的生物大分子动态建模,研究工作越发深入。生物大分子建模的实现,也让人们开始相信整个生命体及所处环境的计算机模拟。
实际上,这一构思已经在实现了--生殖支原体的全细胞模型,所有的生殖支原体的基因、基因产物和已知的代谢相互作用均已在计算机上重建。
单细胞生命体建模的成功预示着我们很快就能实现多细胞有机体的计算机模型。
生物信息学家的定义
纵观整个生物信息学发展史,对于从事生物信息学工作的人并没有清晰的定义。从前,使用计算机分析序列数据的被称为生物信息学家。现如今,生物信息学领域不再只有序列数据,还包括结构生物学上的生物大分子建模数据等等。那现在应如何确切的定义生物信息学家呢?
国际计算生物学学会(The International Society for Computional Biology)根据三个用户类别(生物信息学用户,生物信息科学家和生物信息工程师)发布了生物信息学家在其课程中应具备的核心能力的指南和建议:
基本
使用计算生物学实践所需的当前技术、技能和工具。应用在分子生物学,基因组学,医学和群体遗传学研究背景下的统计研究方法”和“普通生物学知识”对至少一个生物学领域的深入了解,以及对生物学数据生成技术的了解。
拓展
分析问题,并确定和定义适合其解决方案的计算要求(针对生物信息学科学家),以及将数学基础,算法原理和计算机科学理论应用于基于计算机的系统的建模和设计中。