
利用基因流定义微生物种群—PopCOGenT
利用基因流定义微生物种群—PopCOGenT
在微生物中描绘具有生态学意义的种群 (population),对于确定其在环境和宿主相关微生物组中的作用是非常重要的。Martin F. Polz团队从反向生态学角度,开发了能够检测近期横向基因转移事件 (HGT) 的算法—PopCOGenT,可以刻画基因组之间的基因流 (gene flow)。由此将微生物种群定义为基因流较为割裂的 cluster,进而赋予种群以生态学上的意义。
PopCOGenT 的算法逻辑
在了解PopCOGenT 之前,我们要先认识两个概念:反向生态学 (reverse ecology) 和 基因流 (gene flow)。传统的生态学是先看生态上有区别的群体,再研究基因层面上是什么原因造成了它们的生态功能区别。因此,反向生态学就是,直接从研究基因开始,自下而上地推断物种/种群的生态与代谢的特征。在群体遗传学上,基因流(也称基因转移)是指从一个物种的一个种群向另一个种群的基因流动,从而改变种群基因组成。
PopCOGenT就是检测了来同一个种 (species) 的基因组的近期HGT,用有无HGT以及HGT的长度确定基因组 (基因流网络中的节点)之间的基因流关系和大小(基因流网络的边),借此构建网络。将网络上基因流割裂的簇 (网络社区) 划分出来,定义为种群 (population)。因为基因流动频率在种群内高,种群间低,代表了基因流动的隔离,同时基因流将基因功能赋予每个簇,因此也有生态学上的意义,这就实现了反向生态学的方法论--从基因组反推生态功能。
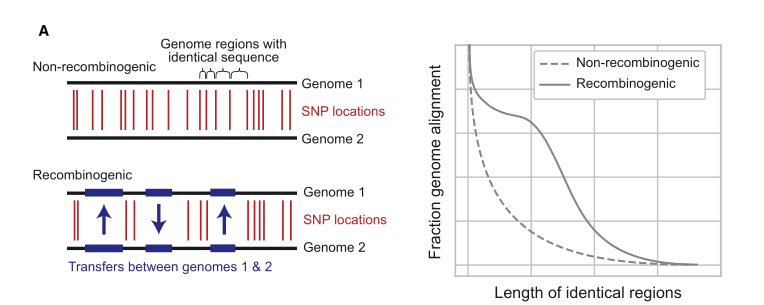
如下图所示,在两个基因组比对 (alignment) 中没有突变的,即序列一致的区域 (region) 称为相同区域 (identical regions)。DNA可能来自垂直遗传或横向转移,横向转移的DNA还来不及积累SNP。因此,只要看到两个基因组之间的相同区域分布显著高于垂直遗传的相同区域分布,那就说明这两个基因组之间有基因转移 (gene Transfer)。 下图中 (右) 虚线是推导出来的没有发生基因转移的基因组alignment对应的identical regions长度的分布,实线是观察到的发生基因转移的基因组的identical regions长度分布。换句话说,就是发生了重组(基因交换)的两个基因组之间的 identical regions 应该比非重组基因组数量上更多、长度上更长,我们将此称为长度偏差 (length bias)。长度偏差可用作为对于基因组同质化 (genome homogenization) 的预测,然后可以构建出最近的基因流网络,并且网络中的社区 (即上述 cluster) 可以被定义为假定的种群。
PopCOGenT在检测基因组之间的基因转移方面是独一无二的,能够检测任何两个菌株共有的所有基因组区域的转移,包括了任何染色体外的可移动遗传物质。
PopCOGenT 的主要应用场景
首先,PopCOGenT 主要功能是应用于相似度很高的基因组(例如,对应16S序列或核糖体蛋白相同或接近相同)之间基因转移的检测及基因流动网络的建立,对这些基因组生成种群结构的假设。给定一组相似度较高的基因组,主要的PopCOGenT模块将识别这些基因组的近期基因流动的关系,并构建原始网络。
那么在基因流动网络中,会发生以下两种情况:
-
在受检测基因组之间没有发现显著的长度偏差,表明这些受检测的基因组可能正在发生evolving clonally 或者与它们发生基因流动的基因组并未在这些受检测的基因组中。
-
在受检测的基因组之间发现显著的长度偏差,这表明发生了基因流动。如果发生了基因的流动,即可在基因流网络中寻找cluster。在这里,我们将原始网络中出现的clusters称为类物种 (species-like) 的基因流单元,之后我们将应用聚类算法,将这些 clusters 分类为以基因流动不连续为特征的种群。
在假设种群后,下一步是识别驱动种群发生差异的基因组区域。PopCOGenT 建立了两个模块识别这些种群特点的alleles和基因。这两个模块的目的是识别种群之间的分化驱动因素,这些种群依然通过基因流动相互连接。
PopCOGenT 主要模块及使用方式
PopCOGenT
利用基因组的一致区域分布的长度偏差来衡量最近的基因组之间的水平基因转移,并且产生种群的预测。
运行PopCOGenT,需要将config.sh文件复制到运行项目所在的当前目录下
(base) [user@server ~]# cp /usr/bio/PopCOGenT-master/src/PopCOGenT/config.sh ./
然后,更改 config .sh 中的 genome_dir 项 ,即基因组数据的目录。注意最后要以/结尾。
如果基因组文件不是以.fasta结尾的,还需要修改 genome_ext,但是建议使用.fasta作扩展名。
进入PopCOGenT的conda环境,运行PopCOGenT
(base) [user@server ~]# conda activate PopCOGenT
(PopCOGenT) [user@server ~]# PopCOGenT.sh ./config.sh
在PopCOGenT 运算结束之后,当前目录下的./output/子目录中生成结果如下:
drwxrwxr-x 2 user user 4096 Apr 14 13:28 proc
drwxrwxr-x 2 user user 4096 Apr 14 13:28 output
drwxrwxr-x 2 user user 737280 Apr 14 13:27 infomap_out
其中,proc目录中的*_length_bias.txt 包含所有基因组之间长度偏差的原始计算。
output中 .graphml 是原始的基因流动网络,可在 Cytoscape 中打开并且进行查看编辑, cluster.tab.txt包含的是最终的种群分配 (assignment)。
如果需要进行网络的聚类,可在 infomap_out 中找到 *.net,利用infomap 将基因流网络中的种群 (population) 进行聚类 (确定cluster)。
具体方法如下:
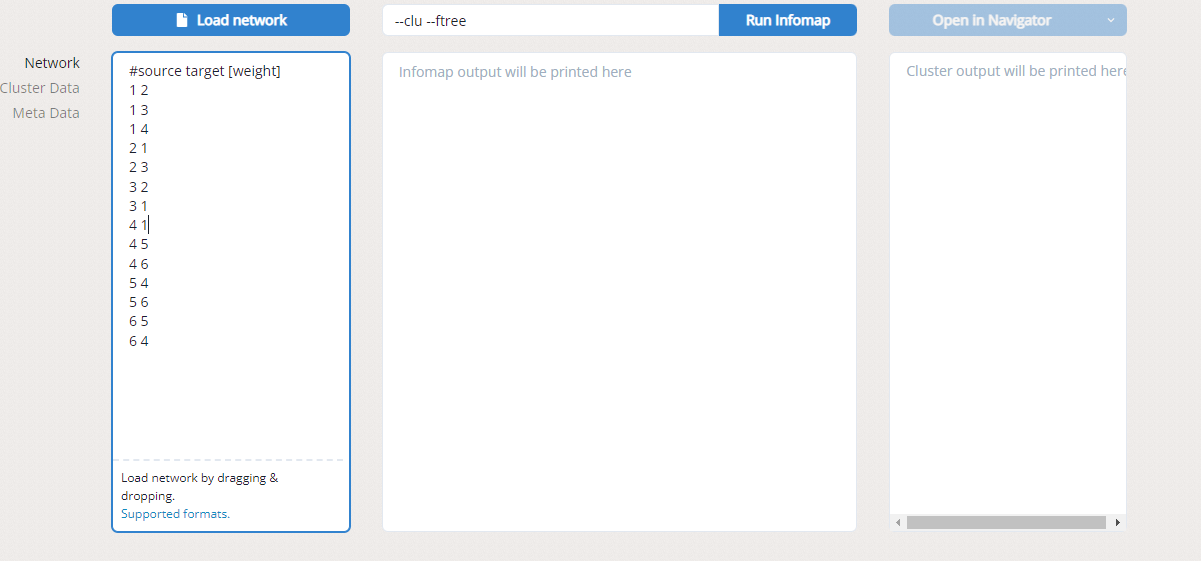
在左侧Load network中导入*.net,之后在Run lnfomap中设置参数,也可以使用图中的默认参数,最后点击Open in Navigator 就可以在网页上显示出网络的聚类关系,使用鼠标可以对网络聚类进行缩放,对网络内具体的基因组进行进一步研究。
Core genome sweep
通过在全基因组比对中识别低多样性、单系(种群内)区域来寻找核心基因的sweep regions。
需要由PopCOGenT 生成的*.cluster.tab.txt 和相关的 *.fasta的基因组文件。还需要指定一个 focus_population,即文件*.cluster.tab.txt的 Cluster_ID 列中的一个。
core_gene_sweeps 的使用方式如下,首先我们需要更改phybreak_parameters.txt, 以下是重要参数说明:
- project_dir : 输出文件目录的绝对路径
- input_contig_dir :输入文件目录的绝对路径
- pop_infile_name :PopCOGenT产生的*.cluster.tab.txt 的路径
- focus_population :*.cluster.tab.txt 的 Cluster_ID。如0.0 、0.1、0.2
- ref_iso :用于alignment的参考基因组,必须只有一个contig
- ref_contig : sequence ID (fasta文件的头的第一个词)
- MUGSY_source: Musgsy的路径
- phyML_loc: phyML的路径
- phyML_properties: phyML的参数,例如-q -m JC69 -f e -c 2 -a 0.022
设置好phybreak_parameters.txt后,运行python 文件1-7进行运算,注意phybreak_parameters.txt 和phybreak1*.py需要在同一目录下运行。运行示例:
shell
(PopCOGenT) [user@server ~]# vim phybreak_parameters.txt
(PopCOGenT) [user@server ~]# python phybreak1.generate_maf.py
(PopCOGenT) [user@server ~]# python phybreak2.maf_to_fasta.py
以上三个文件在
/usr/bio/PopCOGenT-master/src/core_gene_sweeps中。
生成文件如下:
- align/*.core.fasta: 所有alignment 基因组的核心基因的串联
- *.core_sweeps.csv: 核心基因sweeps 的位置
- pop_pi: population 内sweep region 的核苷酸多样性
- Length: sweep region的长度
- Midpoint: sweep region在多个基因组比对的中点坐标
Flexible genome sweep
在微生物基因组中发现和识别种群 ortholog clusters 的工具。
输入文件:
.fasta格式的基因组- PopCOGenT 产生的
*.cluster.tab.txt结果文件
参数设置:
- 复制
*.cluster.tab.txt到./input目录 - 设置参数
config.yaml - Group_to_cluster :
*.cluster.tab.txt中的‘Main_cluster’列,选择你想 cluster 的Group。
配置好后就可以使用以下命令行运行:
(PopCOGenT) [user@server ~]# bash snakemake.sh
snakemake.sh 文件在
/usr/bio/PopCOGenT-master/src/flexible_genome_sweeps中。
生成文件如下: - *.flex_genes.csv :一个csv 文件,其中包含找到的所有flexible ORF 的列表 - orf_id:ORF的名称 - orf_seq: ORF的核苷酸序列 - orf_strand: 表示ORF是正向(1)还是反向(-1) - orf_ortho_cluster: 由mmseqs2生成的ORF直系同源簇的ID - strain:发现ORF的菌株名称
参考文献
Arevalo P, VanInsberghe D, Elsherbini J, Gore J, Polz MF. A Reverse Ecology Approach Based on a Biological Definition of Microbial Populations. Cell. 2019 Aug 8;178(4):820-834.e14. doi: 10.1016/j.cell.2019.06.033.