
fastGEAR算法思想和操作流程
微生物基因组不断受到一系列进化事件的影响,包括突变、基因获得和丢失、基因重排和重组(任何形式的DNA水平转移)。为了研究微生物基因组重组的现象,Pekka Marttinen团队成员研究了一种新算法 fastGEAR,这是一款基于分析序列比对来进行微生物基因组重组推断的软件。与平常使用的其他几款软件不同,比如:BratNextGen和Gubbins,这些软件常用于检测细菌重组的方法专门用于检测来自外源的重组,目的旨在应用于单个世系,但它们在检测世系之间或者物种之间重组的时候就会受到限制。fastGEAR很好的解决了这个问题,它可以用来识别种群遗传结构,推断世系之间或与外部远源物种之间的重组现象。
fastGEAR算法的思想
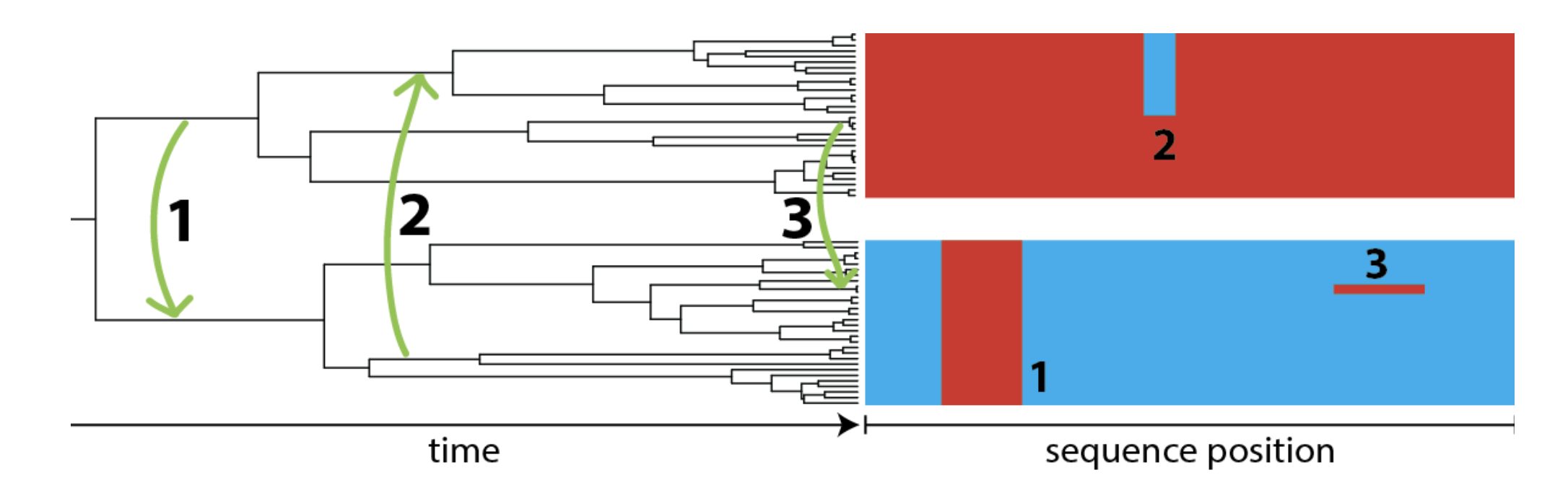
在了解算法之前,我们首先要认识两个概念,recent recombinations(近期重组)和 ancestral recombinations(古老重组)。在下图中可以看到,左边是一棵系统发育树,它有两个类群或者可以说是两个世系,并且被标记成两种颜色。绿色箭头指出了三次重组的流动,右侧是序列比对的结果,颜色代表着祖先的模式。绿色箭头产生的重组,在右侧的比对结果中产生了三个混合的板块,箭头1影响着世系中的所有菌株,此类事件称之为古老重组,箭头2和3影响着世系中的部分菌株,此类事件称之为近期重组。需要我们明白的最重要的一点是,古老重组可能并不是特别古老的,近期重组可能也不是最近才发生的。古老或者近期重组,与我们定义的世系有关。
fastGEAR的主要运行步骤
在输入一个序列比对文件后,fastGEAR会执行四步操作,分别为:世系的鉴定,近期重组的鉴定,古老重组的鉴定和显著性检验。前三步都用到了hidden markov model (HMM),使用BAPS鉴定出遗传聚类,BAPS是一个基因数据聚类的方法,它可以快速找出cluster的数量,但它也有一定的限制,它并不能解释重组相关的信息,比如现有类群A,类群B和拼合的A/B,BAPS将会鉴定出三个不同的类群。
- 世系的鉴定
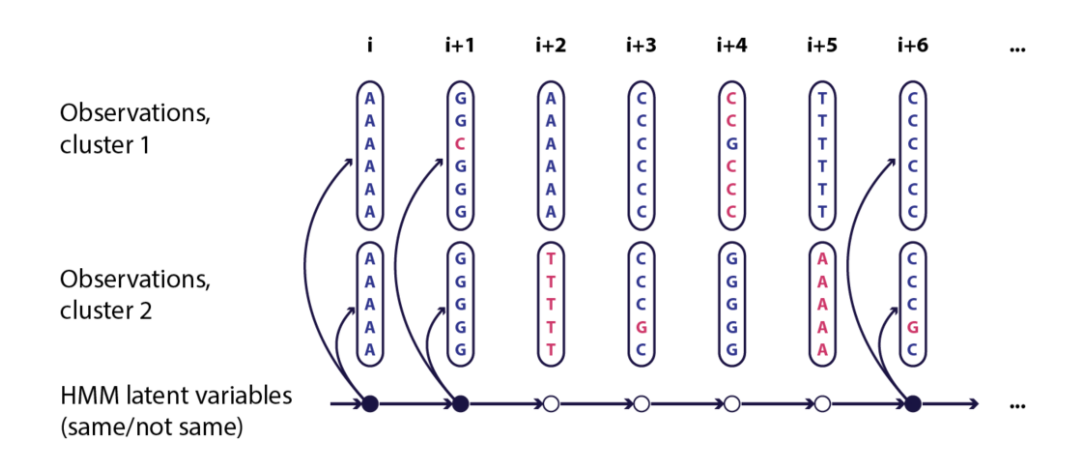
fastGEAR首先使用BAPS聚类,然后鉴定出世系。依据推断出来的clusters,fastGEAR会使用HMM比较每个cluster中的逐个位点(如下图)。我们在图中观察到的是每个cluster中多态位点的核苷酸频率,这一步的主要目的,就是想知道在cluster中的位点是相同的还是不同的,在经过比较之后,fastGEAR会告诉我们答案,如果在两个clusters被鉴定出有50%以上的位点都是相同的,那么这两个clusters就会被鉴定成一个世系。这也表明,cluster是指在基因上有差异的群体,而世系是指在基因的序列比对上至多有50%差异的群体。
- 近期重组的鉴定
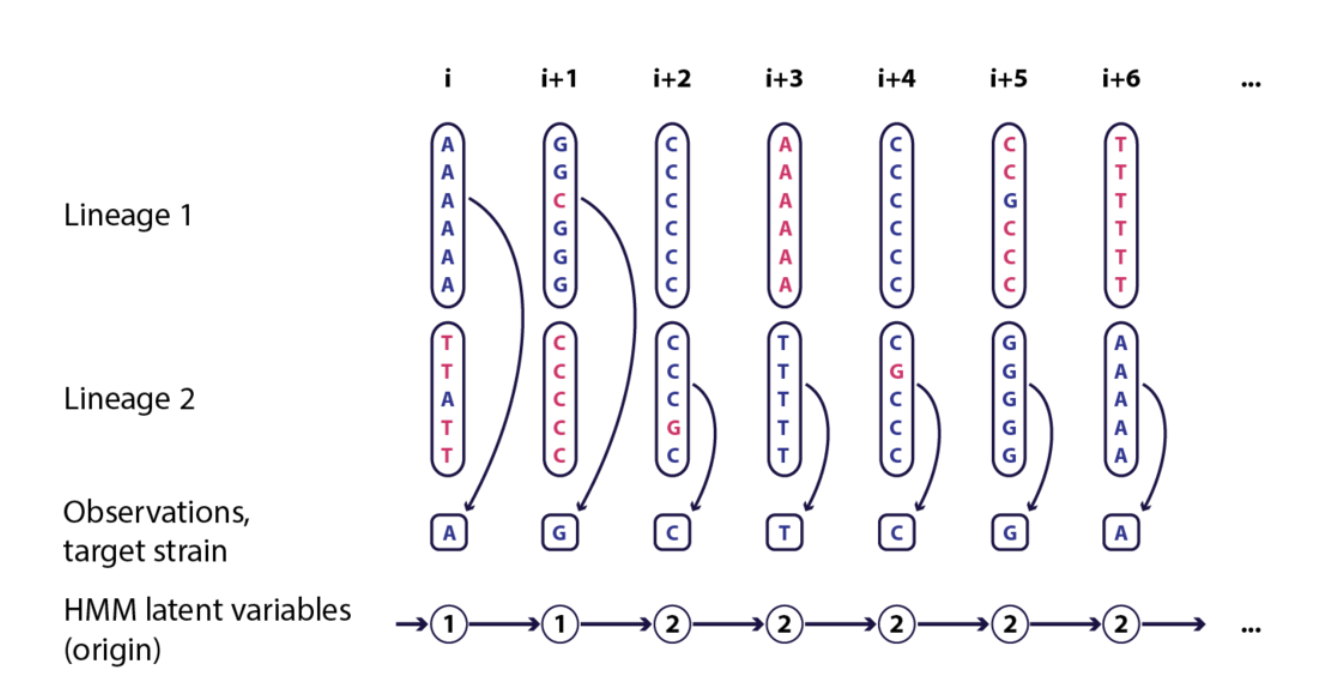
上面用到的是HMM的比较逐个位点方法,在近期重组的鉴定中,使用的是HMM的逐个序列比较的方法。首先,在鉴定出的每个世系和目标序列的比较中,寻找重组。HMM的方法会鉴定出祖先序列(如下图),在祖先序列的基础上,HMM链会比对世系中的每个序列位点,看看这些位点和祖先的相似度是何种状态。如果有些片段是起源于其他世系中的片段,HMM模型会侦测到此片段并将起源定位到此片段,如果分析的片段不是起源于这些比较的序列当中,而且和这些序列存在高度的差异,那么HMM链将会检测到此信号,并将认为它是外部起源。序列中所有来自世系外的东西都可能是潜在的重组。
- 古老重组的鉴定
第三步就是祖先的重组。这和世系的鉴定的原理几乎是一样的,不同的地方就是,世系的鉴定是cluster之间的比较,而这一步是世系之间的比较,并且移除掉了近期重组。通过这种方式,如果HMM模型检测到两个世系中部分比对序列是相同的,那么这两个世系就被认为是古老重组,这是和近期重组不同的地方,并且fastGEAR不检测来自外源的古老重组。
- 显著性检验
这一步主要就是移除掉假阳性的重组。在检测能力比较低时,会出现假阳性的结果,比如在第二步中,出现一些异常值或者离群值,或者在第三步中出现一片有很高的相似度的区域,但不是重组造成的,而是由于自然选择。这两种情况都会造成假阳性的结果。
fastGEAR的安装
在安装fastGEAR之前,我们要先绕一个小弯,因为fastGEAR程序需要在Matlab环境下运行,所以要先安装 Matlab runtime component,下载地址在这里。fastGEAR的下载地址在这里。当以上两个程序安装好之后,另外在Windows中无需配置环境,但需在fastGEAR文件下,下载一个名为fG_input_specs.txt 的文件,下载点这里的example,把下载好的文件放到fastGEAR文件夹下,然后直接使用即可。在Linux中需要在fastGEAR的run_fastGEAR.sh中给出上述安装的Matlab路径才能运行,参数相关的fG_input_specs.txt文件已在Linux版本的软件包中。具体操作细节见下文。
fastGEAR的运行
在Linux中,需在命令行中写入
(base) [user@server ~]$ ./run_fastGEAR.sh <full path to matlab/mcr> <inputfile> <outputfile> <fG_input_specs.txt>
以上是在Linux系统中做的第一步,目前数据分析这一步可以在Linux中进行计算,而对计算的结果做图的工作需要在Window系统中进行。具体在命令行中操作与在Windows中操作一致:
C:\Users\fastGEARpackageWin32bit> plotRecombinations.exe data3_many\alignment-rec.fa.mat 2 2
在Windows系统中,当在电脑上Matlab环境和fastGEAR安装完成之后,输入命令行可以直接使用。同时在运行前还要检查在fastGEAR文件夹下是否存在fG_input_specs.txt,Windows中用法如下:
C:\Users\fastGEARpackageWin32bit> fastGEAR.exe data3_many\alignment-rec.fa data3_many\alignment-rec.fa.mat
C:\Users\fastGEARpackageWin32bit> plotRecombinations.exe data3_many\alignment-rec.fa.mat 2 2
在Linux中最重要的点就是要给出Matlab的路径,在运行fastGEAR的当前目录中必须有fG_input_specs.txt 这个文件,该文件包含的是一些参数,内容如下:
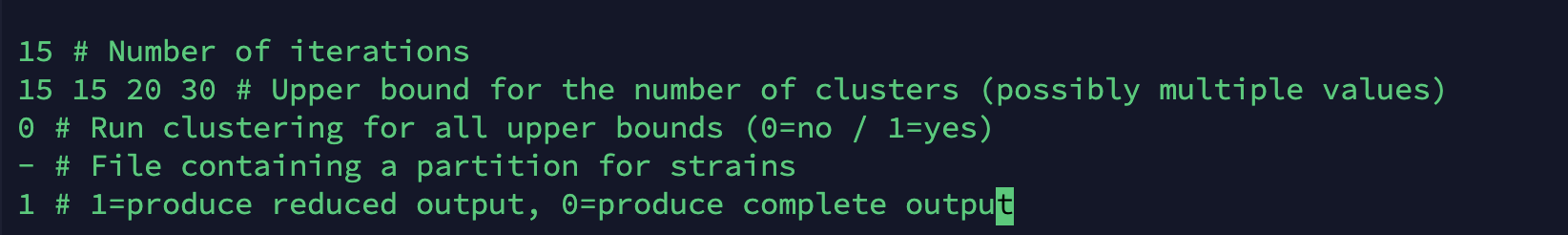
第一行指定迭代学习重组和更新模型参数的迭代次数。收敛可以从屏幕输出监控,它显示了每次迭代的估计参数值。fastGEAR是一种随机算法,也就是当参数值开始振荡,而不是单调递增或递减时,算法可以被认为是收敛的。在用大量数据测试分析之后,fastGEAR团队认为算法通常在10次之内收敛,并给出建议,迭代的次数设置为15次是比较适合分析的。
第二行指定fastGEAR使用的BAPS3聚类算法的初始聚类数量(同时指定cluster数量的上限)。使用每个指定的初始化来运行聚类算法,并返回最可能的聚类。
第三行指定当cluster的数量小于给定的初始数量/上限时,聚类算法是否应该立即停止。这里的基本原理是,如果开始时cluster的数量太少,比如10个,并且您确定了10个cluster,那么应该尝试使用更大的上限。另一方面,如果使用许多不同的初始值运行聚类算法,则需要时间。使用此参数,可以在第二行设置多个初始cluster数量,而且如果找到合适的cluster,就不会浪费更多的时间去聚类。
第四行包含一个文件,这个文件包含了一个预先指定的分区,该分区将用于定义在分析中使用的世系,而不是聚类算法。如果在第四行的位置给出了一个这样的文件,那么我们在第2行和第3行中设定的与聚类算法相关的参数将不再起作用。并且在这个文件中写入的行数应该与你给出的序列的个数一样多。文件的书写格式应该是:每一行都应该有一个正整数,用于指定相应菌株的cluster。在下载的fastGEAR的文件中也给出了一个示例文件./data3_many/data3_partition.txt,可根据自己输入序列的数据,编写指定世系分区的文件。另外,需要注意的是,当手动定义世系时,最大的世系应该有最大的cluster标签。比如,现有大小分别为15,100,30 的三个世系,那么聚类的标签应该是1,3,2。在fastGEAR算法中,这个算法在检测祖先重组时,会自动认为小的世系是重组的。
第五行就是关于结果输出问题,通常情况下,选择默认值1,生成所有相关文件并不是所必须的,并且占用计算资源。
参考文献
Mostowy R, Croucher NJ, Andam CP, Corander J, Hanage WP, Marttinen P. Efficient inference of recent and ancestral recombination within bacterial populations. Mol Biol Evol. 2017 May 1;34(5):1167-1182. doi: 10.1093/molbev/msx066.