
多物种溯祖模型与物种树推断
引言
科学家和公众对稳定可靠的系统发育和完整生命之树的需求每天都在增长。系统发育是进化生物学的一个基本组成部分。它们提供了一个详细的系谱“地图” (genealogical "map"),可应用于各种领域,如生物地理学、分子进化、病原体进化和比较基因组学。近年来,我们推断系统发育的能力急剧增强,不仅是因为高通量DNA测序的技术进步,而且是通过理论上的进步实现的。在过去20年里取得的诸多理论进展中,本文将重点关注多物种溯祖模型 (multispecies coalescent model, MSC) 在系统发育推断中的应用。自20世纪80年代末DNA测序在系统学者中越来越广泛以来,这是系统发育最重要的新方向之一。
溯祖理论的最初观点,也叫做“血统同源性 (identity by descent)”。该概念由S. Wright 和 Molecot在20世纪40年代提出的。但这个前身概念只能够解释2个序列的关系。直到1982年,Kingman J.F.用他杰出的工作构建了整个理论的基本框架,因而推动了这一理论的发展。
当聚合酶链反应在20世纪80年代末被广泛使用时,群体遗传学家和进化生物学家立即开始用DNA序列估计系统发育树。分子系统学当然可以追溯到更远,但单个基因的分子克隆是费力的。随着20世纪70年代末限制性酶的出现及其在DNA多样性中的应用,对物种内的基因树研究首次成为可能。从同工酶和蛋白质多态性向DNA差异的转变对进化生物学界产生了深远的影响,不仅在技术上,而且因为向实证学家和理论家提供的新洞察力。同工酶电泳可以区分特定物种中的不同等位基因,而DNA差异可以测量等位基因之间的进化或遗传距离。这种精度的提高使种群遗传学和系统发育学进入了一个全新的领域。
早期对关系密切的群体和物种的基因树的研究很快发现,来自不同群体的等位基因的基因树并不总是与物种树相一致 (Avise et al., 1987)。这种不一致的最常见的原因之一——当我们在时间上向后观察那些连续的物种形成事件时,等位基因不能完成溯祖合并 (coalesce);或者按时间顺延,在下一个物种形成事件之前,遗传漂变不能足够快地将等位基因“分选”到子代群体。采用时间前向的定义,这种现象被称为不完全谱系分选 (incomplete lineage sorting, ILS),从时间的后向角度看,称为深度溯祖 (deep coalescence) (Maddison, 1997)。基因树-物种树的不一致还可以由其他生物学过程引起,如基因复制、基因渗入 (introgression) 或水平基因转移,但这些都不是群体差异所固有的,因为溯祖过程在所有有限的群体中运作,而其它过程并不总是存在。
Avise 还正式确立了基因树和物种树种之间的区别。物种树的概念是系统发育的同义词,自自达尔文于1859年出版的《物种起源》以来,它一直是进化生物学的基础。然而,正是对自然种群中基因树的实证研究讲清楚了基因树和产生它的物种树之间的区别。在DNA测序的早期,甚至在今天,研究人员经常将基因树视为物种树,或者使用串联方法,假设两者是相同的。虽然基因树和物种树之间的区别已经被审视了几十年,但适应基因树不一致的物种树的估计方法大约从2006年才出现。
Hudson (1983) 导出了三个物种的基因树-物种树不匹配概率。不匹配概率已被用来估计人类与黑猩猩共同祖先的群体大小 (Takahata et al., 1995)。给定物种树时,基因树拓扑的概率 (通常假设一个序列来自一个物种) 得到很多研究者的关注,并开发了自动计算这种概率的算法。从这一系列研究中得到的最著名的结果是存在所谓的异常区 (anomaly zone),即物种树和模型参数在这个区域内,最可能基因树在拓扑上与物种树不同。2003年,带枝长 (溯祖时间) 的任意物种树的全概率分布,即多物种溯祖模型,首次被Rannala和Yang完整的描述。这构成了物种树推理的精确或全似然方法的基础——这些方法直接使用可观察到的DNA序列数据,而不是衍生的摘要数据 (data summaries),如推断的基因树拓扑的集合。
coalesce vi. 联合,合并; coalescence n. 合并,接合,联合; 合生; 聚结; 愈合; coalescent adj. 接合的,结合的,合并的 (也可作名词)。
"The coalescent is the genealogical process of joining lineages when one traces the genealogy of the sample backwards in time".
群体遗传学中,coalescent 翻译为“溯祖”,字面上更能体现上述定义。但在行文中,有时会用“合并”来表达完成溯祖的动作。
多物种溯祖
MSC描述了基因树$G$的概率分布,$G$由两个或多个物种 (或遗传隔离群体) 的基因序列样本构成。该模型是对单个随机交配群体溯祖理论的扩展。因此,我们从描述单一群体溯祖过程开始。
单一群体的溯祖过程
种群遗传学的溯祖理论 (Kingman, 1982; Hudson, 1983; Tajima, 1983) 提供了中性非重组基因座位随机样本的基因系谱历史 (gene genealogical history), 或基因树的概率分布。这个过程通常用单个参数来表示: $$ \theta! = 4 N \mu \quad (1) $$ 其中,$N$是有效群体大小 (理想化的Fisher-Wright模型的群体大小,详见补充材料),而$\mu$是每世代每位点的突变率。虽然在经典的种群遗传学中,$\theta$是用每个基因座位突变率来定义的,但这里使用的每个基因位点突变率在分析基因组序列数据时要方便得多。因此,$\theta$是来自群体的两个随机抽样序列之间,每个位点的杂合性 (heterozygosity) 或平均突变数。
从有效群体大小为$2N$的群体中随机抽出2个序列,它们能在上一代完成溯祖的概率为$\frac{1}{2N}$,因为按照Fisher-Wright模型对群体大小恒定的假设,上一代的群体大小同为$2N$,所以当抽定一个序列后,另一条序列也来自同一祖先的概率即为$\frac{1}{2N}$。如果发生溯祖合并的概率为$\frac{1}{2N}$的话,那么两个序列平均要回溯$2N$代才能完成溯祖合并。因为每世代每位点突变率为$\mu$,所从它们的共同祖先开始,两条序列分别将在每个位点平均积累$(2N)\mu$个突变,那么两条序列之间的位点突变率 为 $2 \times (2N)\mu$,也就是这里的$\theta$。详见补充材料。
原文说有效群体大小为$N$,应该声明这里所说的是双倍体群体。为了让单倍体群体在数学处理上和双倍体群体得到统一,单倍体群体大小为定义为$2N$,双倍体群体大小为定义为$N$。当讨论双倍体群体时,同一个体将携带两个等位基因,所以对于某个基因作为来说群体大小仍为$2N$。
溯祖过程追踪了这些序列从现在一直回溯到过去的系谱历史。样本中的$n$个序列将经历$n-1$个溯祖事件,每次完成溯祖后序列数减少一个,直到整个样本最近的共同祖先 (the most recent common ancestor)。对于一个含有$j$条序列的样本,每一对序列以速率$\frac{2}{\theta}$溯祖 (详见补充材料),对于${j \choose 2}$个所有可能的两两组合有总速率 ${j \choose 2} \frac{2}{\theta}$。两次溯祖之间的等待时间用$t_j$表示 [注意每一次完成溯祖后,谱系 (lineages) 数量或序列数量将减1],$t_j$满足指数分布,并具有概率密度函数: $$ f(t_j | \theta) = {j \choose 2} \frac{2}{\theta} e^{- {j \choose 2} \frac{2}{\theta} t_j} = \frac{j!}{2!(j-2)!} \cdot \frac{2}{\theta} e^{- \frac{j!}{2!(j-2)!} \cdot \frac{2}{\theta} t_j} = \frac{j(j-1)}{\theta} e^{- \frac{j(j-1)}{\theta} t_j} \quad (2) $$
指数分布概率密度函数$f(x) = \lambda! e^{- \lambda! x}$,期望为$\frac{1}{\lambda}$。
指数分布的参数$\lambda$是单位时间内事件发生的平均次数,所以可理解为事件发生的速率。这里的$\lambda = {j \choose 2}\frac{2}{\theta}$,以每个位点的突变率来衡量等待时间时,发生溯祖合并时间的速率。详见补充材料。
所以其数学期望为 $\frac{\theta}{j(j-1)}$。溯祖时间 $\pmb{t} = {t_n,t_{n-1},...,t_2}$ 为独立随机变量,其联合概率密度为: $$ f(\pmb{t}|\theta) = \prod_{j=2}^n f(t_j|\theta) = \prod_{j=2}^n \bigg{[} {j \choose 2} \frac{2}{\theta} e^{-\sum_{j=2}^{n} {j \choose 2} \frac{2}{\theta} t_j} \bigg{]} \quad (3) $$ 其中时间按每个位点的期望突变率重标 (rescaled)。注意,对于DNA序列数据,溯祖时间 (或群体大小) 和突变率不能分别确定,也就是说可估计的参数是$\theta = 4N\mu$,而不是单独确定$N$和$\mu$。
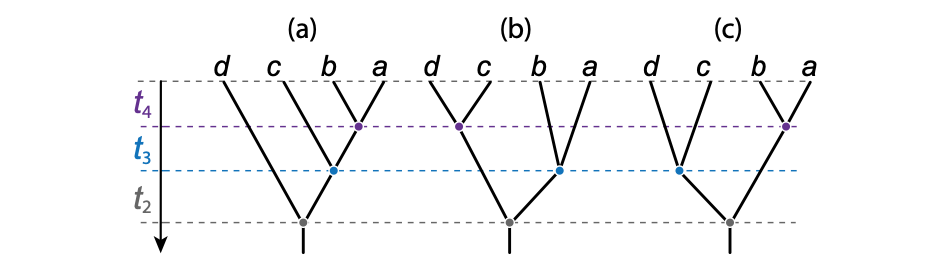
基因树的内部分枝依据年龄排序。其中基因树拓扑$(((a,b),c),d)$ (图1a) 只有一种“标记历史”,而另一个基因树拓扑 $((a,b),(c,d))$ (图1b&c) 有两个不同的“标记历史”,由于内部分枝年龄的排序不同。据公式4,4个序列共有18个“标记历史”,而4个序列的所有可能的基因树共有15个。
溯祖过程也对基因树拓扑结构施加了概率分布。“标记历史 (labelled history)” (Edwards, 1970) 是一种超度量有根二元树 (ultrametric rooted binary tree),树的终端被标记,内部节点按时间或年龄等级排序 (见上图1)。等级次序在完全不对称树 (图1a) 上是完全确定的,但对于对称的树 (图1b&c),可能存在两个或两个以上内部节点的等级次序。在溯祖过程下,所有不同的标记历史都具有相同的概率。Degnan和Salter (2005) 使用了一个不同的术语,将同一“标记历史”的不同等级次序称为同一历史的不同“实例 (instantiations)”。公式3给出了所有可能“标记历史”上的平均溯祖时间的概率密度。$n$个序列可能的“标记历史”数量为: $$ H_n = {n \choose 2} {n-1 \choose 2} ... {2 \choose 2} = \frac{n!(n-1)!}{2^{n-1}} \quad (4) $$ 因为所有“标记历史”在这个过程中都有相同的概率,所以基因树$G = {\pmb{t}, T}$ (这里$G$被一组溯祖时间$\pmb{t}$和一个“标记历史”$T$定义) 的概率为: $$ f(G|\theta) =f(\pmb{t}|\theta) \times \frac{1}{H_n}= f(\pmb{t}|\theta) \times \frac{n!(n-1)!}{2^{n-1}} = \bigg{(}\frac{2}{\theta}\bigg{)}^{n-1} e^{-\frac{2}{\theta}\sum_{j=2}^{n} {j \choose 2} \frac{2}{\theta} t_j} \quad (5) $$
如上图1所示,一定序列的“标记历史”要比可能的二叉有根树拓扑结构多 (甚至多出多个数量级),实际上只有3条序列的情况两者是相等的。主要原因是,”标记历史“考虑内部分枝不同的长度排序,即如图1b&c所示,相同拓扑结构下它们因等待时间 (即分枝长度) 排序差异而不同。这里的等待时间排序,即溯祖时间$\pmb{t}$。换句话说每一个“标记历史”会对应一个溯祖时间$\pmb{t}$。所以$P(G) = P(\pmb{t}|T) P(T)$。这就解释了公式5的由来。
这个概率密度适用于符合中性Fisher-Wright模型的单一随机交配群体的样本,以及其他后代分布可交换的中性模型。它可以在贝叶斯框架中用于使用抽样序列来推断$\theta$。
上述对溯祖的介绍集中在模型下基因树的分布 (拓扑和溯祖时间) 上。溯祖的许多其他方面也可以被研究。此外,基本的中性溯祖模型已经被扩展到允许多个生物学过程,包括随着时间的群体结构变化 (demographic changes)、重组和选择。
多物种溯祖过程
溯祖过程模型已被扩展到多个物种的情况,它们通过系统发育树相关,每个物种中抽样一个或多个序列。有$s$个物种的物种树会有$2s-1$个分枝节点,其中末端节点$s$个,对应于$s$个物种;内部节点$s-1$个,分别于对应$s-1$个祖先物种。因此,在$s$个物种的物种树上的MSC模型有,$s-1$个分歧时间 ($\tau_s$) 和$2s-1$个群体大小参数 ($\theta_s$) (关于群体大小参数详见补充材料)。分歧时间和群体大小都按突变率来按比例缩放,因此$\tau_s$和$\theta_s$都按每个位点的预期突变数来测量。$s=3$个物种 (也可视为群体) 的物种树的参数如图2所示。每个群体在其存续期间内都是一个独立的溯祖过程,群体$i$重标后的 (rescaled) 溯祖率为 $\theta_i = 4N_i \mu$。所有的群体 (除了位于物种树根的群体) 都在由物种分歧时间所决定的有限时间段内存在。
物种分歧时间$\tau_s$,对应于物种树的分枝长度; 有效群体大小$\theta_s$,对应于物种树分枝的宽度。
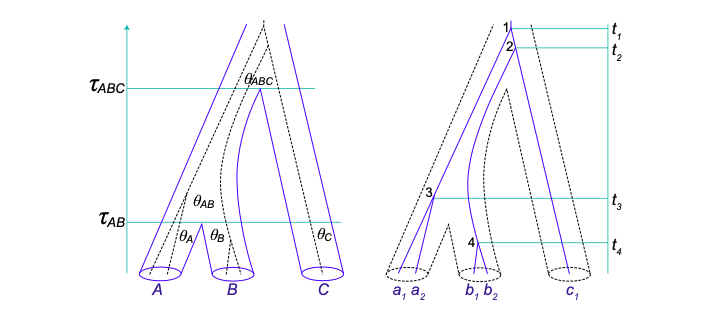
这里的“多物种” 等同于“多群体”。多物种溯祖过程,在物种树的每个分枝上,也就是某个物种的持续时间内,可以用一个单群体的溯祖过程来描述。
物种分歧之后产生的子物种之间,假设是完全隔离的,不存在迁移、重组和渗入。
物种树内基因树的概率密度
给定MSC模型 (具有相关参数的物种系统发育) 下的任意基因树的概率密度由Rannala和Yang 在2003年确定。当考虑物种树时,基因树被认为在基因座位之间是相互独立的。在每个座位上,溯祖过程在物种树上的各群体中也是独立的。因此,我们关注的是:在一个群体中的基因树,比如物种$X$,有亲本物种$P$。用$\tau_X$和$\tau_P$分别表示物种树上两个节点的年龄。$X$可能是一个现存物种,此时$\tau_X = 0$,也可能是一个祖先物种。沿时间轴回溯,用$m$表示在$\tau_X$时进入群体$X$的序列数,用$n$ ($n \geq 1$) 表示在$\tau_P$时 (此时是群体$X$的结束时间),仍保留在群体$X$中的序列数。例如,在图2中,物种$AB$ (年龄为$\tau_{AB}$) 具有祖先物种$ABC$ (年龄为$\tau_{ABC}$)。在图2的基因树中,有$m=3$个序列 (谱系) 进入了物种$AB$,而最终有$n=2$个序列/谱系留在物种$AB$中。溯祖事件之间的$m-n$次溯祖等待时间的概率密度为: $$ \prod_{j=n+1}^m \bigg{[} \frac{2}{\theta} e^{-\frac{j(j-1)}{2} \frac{2}{\theta} t_j} \bigg{]} = \bigg{(}\frac{2}{\theta}\bigg{)}^{m-n} \exp \bigg{{}- \sum_{j=n+1}^m \frac{j(j-1)}{\theta} t_j \bigg{}} \quad (6) $$
溯祖过程是自现存物种延时间轴回溯的,也就是按图所示物种树自下而上的,所以$\tau_A = \tau_B = \tau_C = 0$。
MSC与单种群溯祖的一个重要区别是,在$\tau_P$时,$n \geq 1$条序列/谱系有可能在群体结束时保留。我们必须考虑到$n$个序列在剩余时间内 ($\tau_P - \tau_X - \sum_{j=n+1}^m t_j$) 不能溯祖的概率。无事件发生的概率为: $$ \exp \bigg{{} -\frac{n(n-1)}{\theta} \bigg{(} \tau_P - \tau_X - \sum_{j=n+1}^m t_j \bigg{)} \bigg{}} \quad (7) $$ 如果群体$X$是物种树的根,那么$n=1$,这一项就会消失。将这两个部分相乘,就给出了群体$X$中基因树的概率密度: $$ \bigg{(}\frac{2}{\theta}\bigg{)}^{m-n} \exp \bigg{{} - \sum_{j=n+1}^m \bigg{(} \frac{j(j-1)}{\theta} t_j \bigg{)} -\frac{n(n-1)}{\theta} \bigg{(} \tau_P - \tau_X - \sum_{j=n+1}^m t_j \bigg{)} \bigg{}} \quad (8) $$
泊松过程中,在时间间隔$t$内无事件发生的概率为$e^{-\lambda! t}$,$\lambda$为事件发生的速率。这里$n$条序列的溯祖速率$\lambda = \frac{j(j-1)}{\theta}$ (见补充材料)。所以可得在完成$m-n$次溯祖后剩下的时间内,不发生溯祖的概率,即公式(7)。
该基因座位上整个基因树的概率密度是跨越物种树上所有群体概率的乘积。
例如,给定图2中3个物种的MSC模型,5个采样序列的基因树具有密度 $$ f(G,\pmb{t}|\pmb{\theta}) = \bigg[ e^{-\frac{2}{\theta_A} \tau_{AB}} \bigg] \times! \bigg[\frac{2}{\theta_B} e^{-\frac{2}{\theta_B} t_{4}} \bigg] \times! \bigg[ \frac{2}{\theta_{AB}} e^{-\frac{3 \times! 2}{\theta_{AB}} (t_3 - \tau_{AB})} \cdot! e^{-\frac{2}{\theta_{AB}}(\tau_{ABC} - t_3)} \bigg] \times! \bigg[ \frac{2}{\theta_{ABC}} \cdot! e^{-\frac{3 \times! 2}{\theta_{ABC}} (t_2 - \tau_{ABC})} \times! \frac{2}{\theta_{ABC}} \cdot! e^{-\frac{2}{\theta_{ABC}}(t_1 - t_2)} \bigg] \quad (9) $$
原文中的等式9有误。
第1项,表示对于物种$A$,在$\tau_{AB}$时间内没有溯祖发生的概率。
第2项,表示物种$B$,在$t_4$时间内完成一次溯祖的概率。
第3项,表示物种$AB$,在$t_3 - \tau_{AB}$时间段内完成一次溯祖 (由$m=3$到$n=2$),且在剩下$\tau_{ABC} - t_3$时间段没有溯祖发生的概率。
第4项,表示物种$ABC$,在$t_2 - \tau_{ABC}$时间段内完成一次溯祖 (由$m=3$到$n=2$),且在剩下$t_1 - t_2$时间段有放生1次溯祖的概率。
4对括号中的项分别对应于$A$、$B$、$AB$和$ABC$ 4个物种。当从该物$C$种中只抽样一个序列时,在该物种中是不可能完成溯祖的。
数据中有多个基因座位,所有基因树的概率密度就是基因座位的乘积。该公式允许在不同的座位进行不同的抽样;例如,每个物种的序列数量可能因座位而不同。
溯祖是一个基本的过程,无论物种最近分化的,还是很早分歧的,以及物种是否通过快速的物种形成事件产生 (此时容易产生不完全的谱系分选)。在物种差异相对于群体大小相差很远的情况下 (小群体大差异),物种树将有较长的内部分枝,并且很少有ILS或基因树-物种树的不一致,但这正是MSC模型所预期的。正如Degnan (2018) 所讨论的那样,MSC应该被认为是一个零模型 (null model),而其他生物过程 (如重组、种群结构、基因流等) 可纳入零模型,并衍生出重组MSC模型、群体结构变化MSC模型,和迁移MSC模型、基因渗入MSC模型等等。由于这些模型的复杂性,其中许多尚未实现,但在概念上,它们应该是可能的。
基因树拓扑结构的概率
MSC的另一个令人感兴趣的方面是基于特定物种树和分枝长度的,基因树拓扑的边缘概率,特别是基因树拓扑与物种树匹配的概率。如上所述,“标记历史”在单一群体溯祖过程下具有相同的概率。然而,MSC的情况并非如此。
最简单的情况涉及三个物种$A$、$B$和$C$,有三个序列$(a, b,c)$,从每个物种中取样一个序列。共有三种有根基因树:$G_1 = ((a,b),c)$,$G_2 = ((c,a),b)$,和$G_3 = ((b,c),a)$,这些树拓扑在给定物种树$S = ((A,B),C)$的条件下概率由Hudson在1983年导出。当物种树$S = ((A,B),C)$时,分歧时间为$\tau_{AB}$和$\tau_{ABC}$,祖先群体大小参数为$\theta_{AB}$和$\theta_{ABC}$。序列$a$和序列$b$在祖先群体$AB$内完成溯祖的概率为$1-e^{-x}=1-e^{-(\tau_{ABC}-\tau_{AB}) / \frac{\theta_{AB}}{2}}$;序列$a$和序列$b$在祖先群体$AB$内不能完成溯祖 (在这种情况下,所有三个序列都进入了祖先$ABC$,而这三种基因树发生的概率相等) 的概率为$e^{-x}$。这里,$x = 2(\tau_{ABC} - \tau_{AB}) / \theta_{AB}$被称为以溯祖单位为单位的内部分枝长度:在群体$AB$中一个溯祖单位为$2N_{AB}$个世代,或每位点$\theta_{AB} / 2$个突变。因此,这三种基因树拓扑的概率是 $$ P(G_1|S) = (1-e^{-x}) + \frac{1}{3}e^{-x}, \quad (10) $$
$$ P(G_2|S) = P(G_3|S) = \frac{1}{3}e^{-x}. \quad (11) $$
基因树匹配 (或不匹配) 物种树的概率是 $$ P_{match} = P(G_1|S) = 1- \frac{2}{3}e^{-x}, \quad (12) $$
$$ P_{mismatch} = P(G_2|S) + P(G_3|S) = \frac{2}{3}e^{-x}. \quad (13) $$
当$x \to! \infty$,匹配的概率$P_{match} \to! 1$,不匹配的概率$P_{mismatch} \to! 0$;而当$x \to! 0$,匹配的概率$P_{match} \to! 1/3$,不匹配的概率$P_{mismatch} \to! 2/3$。因此,用基因树最难推断出的物种树是那些内部分枝较短的树。
Degnan和Salter (2005) 开发了计算给定任意数量物种和任意物种树 (从每个物种中抽样一个序列) 的情形下,基因树概率的算法。由于随着物种和序列数量的增加,树拓扑结构数量的爆炸性增长,该算法的计算成本非常昂贵。基因树的概率可用于按最大似然法的物种树估计,将基因树拓扑作为数据处理。这些都是所谓的两步物种树推断方法。在实践中,几乎所有的两步方法都是基于三重有根数或四重无根树,然后组合结果,产生所有物种的物种树估计。
异常区
基因树概率计算中最著名的结果是存在所谓的异常区 (anomaly zone),也就是说:在物种树和参数值的某个区域内,最可能基因树的拓扑不同于物种树的拓扑 (Degnan and Rosenberg, 2006)。3个物种没有异常区,但4个或4个以上物种的有根物种树可能存在异常区。在异常区,使用最高频率的基因树作为物种树估计的“少数服从多数 (majority-vote)”方法将会得到不一致结果。这种基因树被称为异常基因树。异常区的存在是因为溯祖过程在“标记历史”上产生均匀分布,而在有根树拓扑不产生。如图1所示,不对称拓扑只有一个“标记历史”,而对称拓扑可以有两个或多个。这可能会增加对称的基因树拓扑的可能性,即使物种树拓扑是不对称的。
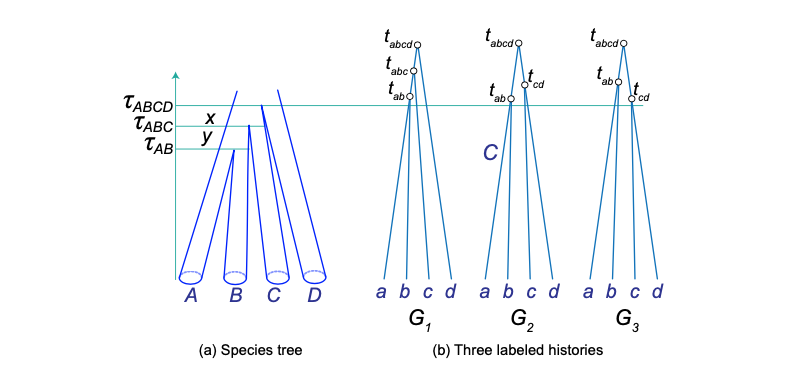
考虑4个物种的情况,有不对称的物种树 $(((A,B),C),D)$ 和4条序列 $(a,b,c,d)$ 的基因树,每个物种抽取一条序列。让物种树中溯祖单位的内部分枝长度为$x = 2(\tau_{ABCD} - \tau_{ABC})/\theta_{ABC}$和$y=2(\tau_{ABC}-\tau_{AB})/\theta_{AB}$。当$x \to 0$,$y \to 0$时,祖先物种$AB$或者$ABC$完成溯祖的概率趋近于$0$,所有溯祖事件将在根部物种$ABCD$发生。在根部物种$ABCD$处的溯祖过程等同于单群体4条序列的溯祖,所以应用公式4,会有18个可能的“标记历史”,每个“标记历史”的概率为$1/18$。其中,12个为完全不对称的 (如图3中的$G_1$),6个为对称的 (如图3中的$G_2$和$G_3$)。每个不对称“标记历史”都对应于一个唯一的树拓扑,因为只有一种可能的方法来对内部节点进行排序。6个对称的“标记历史”形成3对,每对对应于一个具有两个可能的节点顺序的树拓扑 (例如图3中的$G_2$和$G_3$)。因此,每个对称有根树拓扑的概率为$1/18+1/18=2/18$,而每个不对称根树拓扑的概率为$1/18$。当分枝长度$x$和$y$非零但非常小时,对称而不匹配的基因树 (图3中对应于$G_2$和$G_3$) 仍然可能比不对称但匹配的基因树$G_1$有更高频率,即使达不到其概率的两倍。因此,频率最高的基因树将具有一个与物种树不匹配的对称拓扑结构,而这种物种树拓扑和分枝长度的组合就在异常区。
原文第8页,第2-3行:“Of these, 12 are fully asymmetrical (such as labelled histories G2 and G3 in figure 3) and 6 are symmetrical (such as labelled history G1 in figure 3)” 是错误的。
异常区已被证明会影响来自蜥蜴的经验数据集,还有不会飞的鸟类、长臂猿和非洲蚊子。异常区可以通过使用bpp等贝叶斯推理程序估计MSC模型中的参数来识别,然后模拟基因树使用这些参数来估计基因树概率,以确认最可能的基因树与物种树不匹配。
虽然众所周知,内部分枝非常短的物种系统发育很难从新获得,但异常区的重要性可能在文献中被夸大了。请注意,异常区只是简单的“少数服从多数”方法的不一致区。其他方法在异常区域可能不一致,也可能一致。特别是,基于序列数据似然函数的方法,包括最大似然和贝叶斯方法,在异常区内外都是一致的;事实上,它们在整个物种树空间上是一致的。
物种树推断方法
MSC为开发物种树推断的有参数多基因座位统计方法提供了一个框架。这种方法允许基因树由于ILS而不同于物种树,并提供对祖先群体结构统计参数的估计。由于MSC在所有有限种群中适用,因此它是物种树推断的典型模型。我们首先描述为物种树推断而开发的最大似然方法和贝叶斯方法。这些方法通常被称为全似然方法,因为它们使用了一个精确的似然函数。全似然方法被称为具有一致性和效率的最优统计特性。然后我们介绍两种最广泛使用的近似方法:MP-EST和ASTRAL。这些程序是“超级树”方法的例子,它们通过合并较小的树的估计来推断较大的树。其中MP-EST使用伪似然来近似MSC,而ASTRAL使用一个简单的启发式算法,当基因树在MSC下出现时,可以提供统计上一致的估计。启发式方法的统计性质通常只能通过计算机模拟来研究。其它近似方法的介绍见Yang and Rannala (2014), Edwards (2016) 和 Xu and Yang (2016)。
接下来,我们考虑另一类近似方法,即所谓的串联法。这些方法将所有的座位串联成一个单一的序列矩阵,是“超级基因”或“超级矩阵”方法的例子,隐式地假设没有ILS。Gatesy and Springer (2013) 和 Edwards et al. (2016) 综述了关于物种树推理的两步法和串联法的相对优缺点的广泛讨论。我们简要地总结了使用串联的近似推断方法可能出现的几个问题。最后,我们讨论了对基于MSC的两步近似推理方法和全似然方法的一些批评。
最大似然法
用$X_i$表示在座位$i$上得到的序列比对,其中$i = 1,2,...,L$,向量$\pmb{X} = {X_i}$表示所有$L$各座位上的序列比对。然后用$\pmb{\theta}k$表示MSC在物种树$S_k$上的模型参数 ($\theta_s$和$\tau_s$),用$G_i$表示座位$i$上的基因树。与传统系统发育方法的主要区别在于,基因树是未被观察到的随机变量,其分布由MSC模型指定 (Rannala and Yang, 2003)。例如,物种树估计的最大似然方法使以下似然函数最大化 $$ f(\pmb{X} | S_k, \pmb{\theta}_k) = \prod_k) f(X_i | G_i) d G_i \bigg] \quad (14) $$ 其中$f(G_i|S_k, \pmb{\theta}_k)$为基因树$G_i$的在MSC模型下概率密度,$f(X_i | G_i)$为座位$i$上序列比对的概率 (或系统发育似然)。对基因树$G_i$的积分,代表了座位上所有可能的基因树拓扑 (“标记历史”) 的总和,以及每个基因树上溯祖时间的积分。在这个公式中,基因树$G_i$是未被观察到的随机变量 (称为潜在变量),物种树和MSC参数的似然函数必须平均每个座位上所有可能的基因树。}^{L} \bigg[ \int! f(G_i|S_k, \pmb{\theta
使对数似然$\ell = \mathrm{log} f(\pmb{X} | S_k, \pmb{\theta}_k)$最大化的$S_k$和$\pmb{\theta}_k$即是最大似然物种树,和该物种树上的MSC模型参数的最大似然估计。注意,MSC模型下概率密度$f(G_i|S_k, \pmb{\theta}_k)$和系统发育似然$f(X_i | G_i)$是容易计算的。真正的困难在于对每个基因座位上的可能基因树进行平均,因为可能的基因树数量是巨大的,并且每个具有$n$个序列的基因座位上的每个基因树,其溯祖时间的积分是$n-1$维的。基于MSC,唯一实现ML法的软件是3s,它枚举了基因树拓扑并使用数值积分 (高斯四度) 来计算积分。它的应用被限制在三个物种和三个序列,但可以容纳数万个座位。
贝叶斯推断
在贝叶斯方法中,我们为所有可能的物种树指定一个先验概率分布,而对于每个物种树,我们为MSC模型的参数 ($θ_s$和$\tau_s$) 指定一个先验分布。设 $f(S_k)$是物种树$S_k$的先验概率。这可以是在有根物种树或“标记历史”排序树上均匀分布,此外我们通常在给定物种树$f(\pmb{\theta}k|S_k)$的MSC模型参数上分配伽马或逆伽马先验分布。逆伽马先验与$\theta$共轭,允许$\theta_s$被解析积分。由于减少了参数空间,这可以稍微改善树搜索过程中马尔可夫链蒙特卡洛 (MCMC) 的混合。通常,在根的年龄 ($\tau_0$) 上指定一个伽马或逆伽马先验,而其他节点的年龄则可以由狄利克雷分布或出生-死亡过程模型来指定。贝叶斯计算通过MCMC算法实现,该算法从物种树和基因树的联合条件分布 (联合后验) 中生成一个样本 $$ f(S_k,\pmb{\theta}_k,{G_i}|\pmb{X}) \propto! f(S_k)f(\pmb{\theta}_k|S_k)\prod_k)f(X_i|G_i)]. \quad (15) $$ 与公式14相比,基因树上的积分消失了,并通过MCMC进行数值积分。换句话说,MCMC被用于穿越基因树的联合空间,以及物种树和MSC参数。MCMC访问每个物种树的频率是对该物种树的后验概率的估计。贝叶斯方法的第一个实现是Best程序,它使用了MrBayes程序的基因树的后验样本,并对基因树密度进行了修正,因为贝叶斯的基因树不是基于MSC先验产生的。后来的实现软件直接处理MSC和序列比对,而不是处理MrBayes输出,特别是在程序StarBeast和bpp中。树搜索中的分支交换算法,如最近邻居交换 (NNI)、亚树修剪和嫁接 (SPR) 等,也被调整以适应在MCMC过程中,对物种树做变动。}^{L}[f(G_i|S_k,\pmb{\theta
MCMC算法对物种树推断的最大挑战似乎是物种树和基因树之间的约束。如果当所有基因座位的基因树固定时,如果物种树发生改变,当前的基因树可能会对物种树施加极其严格的限制。考虑改变了两个物种 (或分枝) $A$和$B$之间分歧时间$\tau_{AB}$的简单变动。在溯祖模型中,序列分歧时间必须早于物种分歧时间,即$t_{ab} > \tau_{AB}$,这个约束适用于每个座位的两个物种 (或分枝) $A$和$B$的每一对序列。换句话说,当前的基因树为$\tau_{AB}$提供了一个最大界,这是贯穿所有座位的最小$t_{ab}$。如果在数据集中有数千个座位,并且在每个座位上都有许多来自A和B的序列,那么$\tau_{AB}$的当前值通常与这个边界几乎相同,而不可能变得更大,算法本质上是被卡住了。一个似乎很好的算法是bpp实现的橡胶绑定算法,它能够识别受$\tau_{AB}$改变而受影响的基因树上的节点,然后在$\tau_{AB}$改变的同时,修改这些基因树节点的年龄。这就橡胶带上的标记点在两端固定时,橡胶带从中间的一个点拉到一端。这一举措已经被移植到StarBeast中。当物种树的拓扑结构发生变化时,物种树和基因树之间的类似的协调变化似乎改善了MCMC的混合。这些智能的MCMC变动使得分析具有超过1万个位点的大型数据集成为可能。在计算和混合效率方面显然需要进一步改进,因为对于贝叶斯MCMC程序能够处理的量来说,真实的数据集通常是非常大的。
近似物种树推断方法
下一代测序技术目前正在以惊人的速度进步,使数百个个体和物种能够获得密集的基因组序列。这大量的新数据推动了对推断物种树方法的需求,这些方法可实际应用于数千个座位和数百或数千个序列。基于模型的物种树估计方法,如上述贝叶斯推断程序,是计算密集型的,并将落后于许多当代测序项目的需求。因此,提出了许多启发式 (或近似) 物种树推断的方法,使用各种快捷方式和启发式近似来提高大型数据集的计算效率。所讨论的一些启发式方法 (MP-EST和ASTRAL) 明确地尝试来适应ILS,而其他方法 (串联法) 则没有这样做。
一类近似物种树推断方法——超树方法 (supertree methods) (如MP-EST和ASTRAL) 采用两步法,从单个基因座位序列比对的系统发育分析中估计基因树,然后将基因树视为观测数据。第二类近似的物种树推断方法——超矩阵或超基因方法 (supermatrix / super-gene methods), 将所有座位串联成一个序列,假设超矩阵的基因树与物种树匹配。在一个基因树与物种树相匹配的假设下,这种方法通常应用一种标准的最大似然法或贝叶斯系统发育方法。
两步超树方法比全似然方法容易实现得多,也是在MSC下推断物种树的早期方法之一。其中一些方法使用具有分枝长度 (节点年龄) 的估计基因树拓扑,如在STEM程序中实现的最大树方法 (maximum tree method)。一个严重的问题是,该方法没有考虑到估计树拓扑和溯祖时间中的抽样误差。尤其,溯祖时间可以对物种树的推断产生重大影响:例如,如果两个物种 (或分枝) $A$和$B$的两个序列在任何座位是相同的,即$t_{ab}=0$,那么物种分歧时间$\tau_{AB}=0$。这种对物种分歧时间的极端估计将影响物种树拓扑结构的推断。其他的两步法使用估计的基因树拓扑作为数据,忽略分枝长度或溯祖时间。这些方法从数据中使用的信息较少,因此受系统发育重建误差的影响也较小。它们通常在无根基因树上有效,这些无根基因树的估计不需要有分子钟的假设。这些仅基于拓扑的方法比基于分枝长度的基因树的方法更成功。然而,许多两步法的统计性能都很差,而且一些方法的精度 (如STEM) 甚至随着座位数量的增加而减小。基于串联的超基因法依赖于现有的单基因座位系统发育推理方法的直接应用,因此应用起来很简单。然而,由MSC和其他过程导致的基因树和物种树 (在分枝长度和拓扑方面) 之间的差异可能会导致这些方法在统计上不一致。
MP-EST:最大伪似然估计
最大伪似然估计 (maximum pseudo-likelihood estimation, MP-EST) 方法是一种基于物种三联 (species triplets) 的两步估计方法。对于含有$s$个物种的树,提取所有的$s(s-1)(s-2)/6$个有根三联种子树 (每个树由3个物种组成) 来构建似然函数。每个有根三联种子树中的单个内部分枝长度,是原始$s$个种的树中一个或多个内部分枝长度的总和。输入到程序的数据是使用最大似然或贝叶斯推序推断的有根基因树拓扑。给定物种亚树的每个三联体基因树拓扑的数量遵循三项式分布,概率由MSC模型决定 (公式10)。三联体的基因树拓扑的概率与物种子树和基因座位相乘。这是一个伪似然函数,因为它忽略了三联体子树是不独立的事实。利用启发式搜索算法最大化伪似然值以推断物种树。MP-EST方法可能会遭受信息丢失,因为它忽略了基因树中的分枝长度,也忽略了基因树重建中的系统发育误差;所有超树方法都是如此。
注意,MP-EST方法的理论在公式10中给出。通过给出这三种基因树的概率,我们可以找到最常见的基因树拓扑结构,即物种树的估计,并估计物种树的内部分枝长度 ($x$)。如果基因树已知且没有误差,该方法显然是一致的:当基因座位或基因树的数量接近无穷大时,找到正确物种树拓扑的概率接近1。此外,在这种三联种和有根基因树中,Yang (2002) 表明,最有可能估计的基因树拓扑是与物种树匹配的,尽管系统发育重构误差具有推断基因树-种树不匹配概率的影响。因此,当使用估计的基因树拓扑来估计物种树时,MP-EST方法将是一致的。然而,由于系统发育误差扭曲了基因树的概率和膨胀了基因树-物种树不一致性,物种树的内部分枝长度被不一致的估计 (并被低估)。
ASTRAL:准确物种树算法
ASTRAL是另一个两步法程序,以使用最大似然系统发育程序RAxML推断的无根基因树作为输入。背后的方法是基于四联种 (quartets),即有四个物种的四个序列。然后,符合最多四联种基因树的树被选择为物种树。如果一个物种有多个序列,则对每个物种的一个序列被抽样以组成四联种。使用无根四联种进行优化,而不是找到与最多完整基因树兼容 (“少数服从多数”原则) 的物种树的动机是,四物种的无根物种树没有异常基因树。
ASTRAL本质上使用了基因树拓扑的ML估计作为推断的摘要统计量。它不使用来自基因树的分枝长度信息。仅使用基因树拓扑就可以识别物种树拓扑,以及溯祖单位中的内部分枝长度,但MSC模型中的其他参数则不可识别。注意,虽然该方法被认为是一致的,但一致性的证明依赖于假设基因树没有误差,而系统发育重建误差的影响通常是未知的,尽管这有时是可使用计算机模拟来评估。
串联法
使用多座位序列数据来推断物种树的一种简单方法是将跨座位的序列串联起来,然后使用“超级基因”序列作为物种树的估计来推断单棵树。这隐含地假设所有基因座位共享相同的拓扑和分枝长度。长期以来,系统学家一直在争论是否将不同基因结合成单一分析的问题。从统计的角度来看,分析异构数据的一种标准方法是进行适应异质性的组合分析。然而,在MSC模型的开发和实施之前,没有正式的统计方法允许多个基因被组合,同时尊重他们不同的历史。当物种树很容易,有长的内部分枝和小的群体大小,人们期望很少的深度溯祖或不完全的谱系分选。在这种情况下,串联和溯祖方法有望产生相同的物种拓扑结构。然而,当物种树具有挑战性时,内部短分枝和群体规模大,串联可能不一致,并可能收敛到不正确的物种树拓扑。
即使基因树共享拓扑结构,由于溯祖的波动,它们也可能有不同的分枝 (溯祖时间)。在这种情况下,串联可能导致主要进化参数的估计偏差,而溯祖方法 (应用于序列比对的全似然方法) 适应可变溯祖时间,会提供可靠的估计。最近对不同系统基因组数据集的贝叶斯分析 (Jiang et al., 2019) 表明:(i) 基因树的异质性是真实和丰富的,即使考虑了基因树误差;(ii) 基因树拓扑一致的假设在几乎所有的数据集中都可以被拒绝;(iii) MSC模型比串联法更适合系统基因组数据集。串联仍然是一种广泛使用的方法,特别是在对最近测序的基因组的比较分析中,主要是因为它的简单性和较低的计算成本。随着基于MSC的物种树推断算法的改进,以及对适应物种溯祖并过程重要性的更广泛的认识,这种情况可能会改变。
对多物种溯祖模型物种树推断方法的批评
基于MSC的物种树树推断方法假设没有基因座位内重组。Gatesy和Springer (2013) 正确地指出,当转录组数据中的多个外显子串联成一个基因或座位时,外显子可能沿着染色体跨越很长距离;这种混合串联-溯祖方法可能导致违反MSC模型。基于经验计算,Springer和Gatesy (2016) 预测,一个典型物种辐射 (species radiation) 中的非重组单元足够短,足以违反MSC没有重组的假设。然而,他们的计算并没有考虑在样本中只有一个序列的时间段内发生的重组事件与MSC假设一致的情形。此外,模拟数据表明,仅在极端ILS水平下,MSC方法的座位内重组才会成为一个问题。没有重组的假设对于串联法,比两步法更有问题,因为串联法假设所有基因中的所有位点都有相同的系谱历史,这几乎肯定是违反的。
如上所述,两步法将估计的基因树作为数据,不考虑系统发育误差;这可能导致两步法低估物种树上的内部分枝,并通过放大基因树与物种树的不一致来夸大ILS的重要性。这种批评特别适用于两步法,因为全似然方法通过系统发育似然函数正确地适应基因树误差 (公式14和15)。最近的努力利用自展分析和其他基因树不确定性的评价措施来纠正两步法中的系统发育的不确定性,可能有助于减少系统发育误差的影响。上述讨论在很大程度上适用于密切相关物种的浅层系统发育。对于涉及远缘物种的深层系统发育,一整套影响系统发育分析的复杂因素将影响物种树的推断,包括对分子时钟的违背、跨基因座位和谱系的替代过程的异质性。这些因素与深层溯祖一起,使通过古老辐射物种形成事件产生物种的深层系统发育推断成为一项非常具有挑战性的任务。我们注意到,违背模型是系统发育中的一个共同特征,一个错误指定的模型是否仍然有用可能取决于许多因素,包括模型对分析的影响。更复杂的模型,特别是解释迁移或渗透的MSC模型,可能比没有基因流的基本MSC更好,在存在深度溯祖和渗透的复杂情况下可能导致更好的推断。
未来的挑战
多物种溯祖模型的发展是分子系统发育学的一个重大进展。该模型适应了整个基因组的系谱历史的波动,并为使用来自密切相关物种的基因组序列数据进行推断提供了一个自然的框架,弥合了系统发育学和种群遗传学之间的差距。MSC是解决系统发育基因组学和种群基因组学中许多令人兴奋的推断问题的基础,包括对祖先群体大小的估计和对古老杂交事件的推断,甚至是那些涉及已经灭绝的物种的杂交事件。
目前,MSC模型全似然法的实现,主要以MCMC算法的形式,涉及密集的计算。随着数据大小的增加 (例如物种的数量,每个序列的位点数量,每个座位的序列数量和座位的数量),每次MCMC迭代都需要更多的计算工作。此外,MCMC混合存在衰退现象,因此需要更多的MCMC迭代,以产生可接受的有效样本量。目前的大多数MCMC实现对于具有数千个座位规模的数据集的计算都是不可行的,尽管bpp提出对基因树和物种树的协调变动,可以分析超过10,000座位的数据集。进一步改进算法的计算和混合效率是非常值得的。
基因组序列数据的爆炸性增长意味着近似方法或启发式方法将继续在数据分析中发挥重要作用。目前的两步法似乎只利用基因组数据集的一小部分信息,特别是在分析浅系统发育密切相关的物种,因此MSC模型中的许多参数是两步法无法识别的,即使对于物种树拓扑也是如此。发展统计上更有效的启发式方法应该是未来研究的优先事项。
对于有远亲关系的物种,分子钟假设可能会受到严重的违背。尽管人们可以采用系统发育学中开发的放松时钟模型来适应对时钟的违背,但速率变化意味着基因树中的一些时间信息被侵蚀。在具有放松时钟的MSC下,全似然方法与使用无根基因树拓扑和忽略基因树分枝长度中的时间信息的启发式方法相比,还有待观察。
我们预计,在MSC模型中适应跨物种基因流将成为未来几年的一个研究热点。许多最近的实证研究表明,跨物种基因流在动物和植物中很常见,甚至跨越生命树。MSC模型可以被扩展,以适应跨物种的基因流动。目前已经开发出了两种这样的模型。MSC-迁移模型,更广为人知的是迁移隔离 (isolation-with-migration, IM) 模型,假设持续迁移,物种每一代以一定的速度交换移民。该模型与群体细分的群体遗传学模型相似,但在IM模型下,群体具有分枝顺序和分歧时间的系统发育历史。IM模型下基因树的概率密度由Hey and Nielsen (2004) 和Hey (2010) 给出。结合基因渗入的MSC (MSci) 模型,也被称为多物种网络溯祖 (MSNC) 模型,假设情景渗入/杂交;换句话说,渗入发生在过去的某个时间点。模型中的重要参数包括渗入时间和概率。Yu et al., (2014) 给出了MSci模型下的基因树概率密度。贝叶斯MCMC的实现包括针对IM模型的IMa3,以及针对MSci模型的StarBeast和PhyloNet。这些程序涉及到昂贵的计算,而且对于较大的数据集是不可行的,比如说,它们有超过200个座位。与此同时,这些模型的复杂性意味着,具有数千个座位的大型数据集可能需要获得可靠的参数估计。在IM模型的情况下,MCMC平均在每个座位的一个巨大的系谱历史空间上求平均,其中包括迁移事件的数量和方向。在高迁移速率下,这个空间实际上是无限的,似然表面在这个空间上几乎是平坦的,因为序列似然取决于基因树的拓扑结构和分歧时间,而不依赖于迁移事件。对于MSci模型,一个主要的绊脚石是物种树或网络和基因树之间的约束,就像在简单的MSC模型中一样。最近开发的bbp,可实现模型参数如物种分歧或杂交时间与基因树之间协调变动,这使得分析10,000多个座位的数据成为可能,但目前模型是固定的,由用户指定的渗入事件的数量和方向。MCMC允许在不同MSci模型之间移动的动议尚未实施。迫切需要提高全似然方法的计算效率。
几种启发式的方法已经被开发来检测跨物种基因流和估计渗入概率。有些软件采用两步法并使用估计的基因树拓扑,如SNaQ。其他的则使用多座位序列数据的其他摘要方法,如三物种或四物种的简约信息位点模式,包括流行的ABBB-BABA检验和HyDe程序。这些方法没有使用基因树的分枝长度信息,尽管最近的 Hibbins and Hahn (2019) 的启发式方法确实尝试了使用分枝长度信息。它们可以估计物种树上溯祖单元的深入概率和内部分枝长度,但模型中的其他参数无法识别。此外,许多基因渗入场景是无法识别的,也不能使用这些方法检测到。在渗入参数可识别的情况下,两步法似乎提供了与完全似然方法相似精度的估计。在未来几年,开发统计高效的启发式方法应该是高优先事项。
未来研究MSci模型的另一个重要途径是它们的可识别性。这些数据可以是基因树拓扑 (对于两步启发式方法) 或多座位序列比对 (对于全似然方法)。可识别性可能涉及不同的渗入模型 (假设不同数量的渗入事件,或假设涉及不同物种的渗入事件),也可能涉及给定的渗入模型中的参数 (包括物种分歧时间、群体规模和深入概率)。一些可识别性问题可以通过在两步法中使用更多信息丰富的摘要统计数据来解决,但这可能会使启发式估计器的推导更加困难。
物种树推断是一个困难的统计问题,特别是当纳入诸如基因渗入等因素时。MSC是一个将种群遗传学与进化历史联系起来的模型,因此它是物种树推理问题的核心。我们期望,在可预见的未来,有效和准确地推断物种树的目标将仍然是系统发育推断学科的核心。虽然在过去的二十年里已经取得了很大的进展,但仍存在许多具有挑战性的问题。
全文引自:Bruce Rannala, Scott V. Edwards, Adam Leaché, and Ziheng Yang (2020). The Multi- species Coalescent Model and Species Tree Inference. In Scornavacca, C., Delsuc, F., and Galtier, N., editors, Phylogenetics in the Genomic Era, chapter No. 3.3, pp. 3.3:1–3.3:21.
补充材料
Fisher-Wright模型
Fisher-Wright模型 (Fisher, 1930; Wright, 1931) 对群体的繁殖条件做了如下理想化的假设:1. 世代是离散时代,并且世代不重叠;2. 群体是单倍体或两个子群体 (雌性与雄性,即二倍体);3. 群体大小在每一代是常数;4. 所有个体的适合度相同;5. 群体没有地域结构和社会结构,下一代的基因是从前一代基因库中有放回的随机抽取的;6. 群体中的基因没有重组。
物种/群体内溯祖模型
两条序列的溯祖
假设现在从一个满足Fisher-Wright模型的双倍体群体 (群体大小为$N$) 中随机抽取某一个基因座位的序列$2$条 (注意,当考虑某座位上的基因时,群体大小就成为$2N$了)。它们必定来自于曾经的存在的某个最近的共同祖先 (most recent common ancestor, MRCA),假如我们现在如果沿着时间向后去寻找它们的MRCA,那么要经过多少个世代才能找到呢?
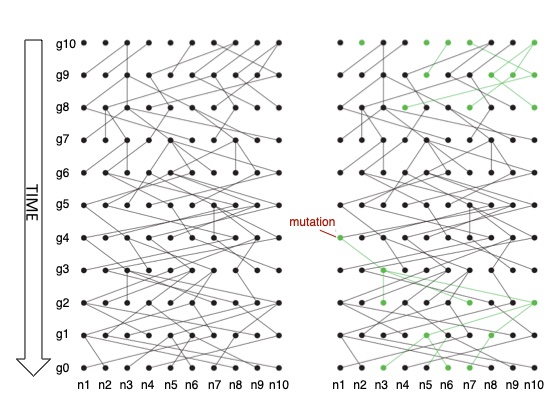
回想一下这2条序列的抽取过程,应该是这样的:首先从当前世代中随机抽取1条序列 (1条序列的溯祖是没有意义的)。接着我们再随机抽取第2条序列,此时溯祖事件就会发生。它们可能在上一代就合并了 (如附图1中g0代的n3与n6),也可能在较久远的某一代合并 (如附图1中g0代的n1与n3)。那么在上一代就合并的概率是多少呢?因为满足Fisher-Wright模型的群体有恒定的大小,所以上一代的群体大小同为$2N$,那么当选定1条序列后,第2条选中和第1条同祖先的那条序列的概率就$\frac{1}{2N}$。换句话说,也就是在上一代中选中特定某1条祖先序列的概率。有了在上一代合并的概率$\frac{1}{2N}$,其逆事件“在上$1$代不能合并”的概率即为$1-\frac{1}{2N}$。那么在上$2$代不能合并的概率就是$(1- \frac{1}{2N})^2$。
将这个公式一般化,也就是在上$i$代不能完成溯祖的概率为:$P(T_2^{\prime} > i) = (1 - \frac{1}{2N})^i$,其中$T_2^{\prime}$表示2条序列完成溯祖合并的等待时间 (注意,这里的等待时间是用世代数表示的),让$i=2$,$T_2^{\prime} > 2$的概率也就是溯祖合并事件发生在上$2$代以上某一代的概率,也就是“在上$2$代不能合并“的概率。再进一步的问:刚好在第$i$代完成溯祖合并的概率是多少?这个事件实际上蕴含着“在上$i$代不能完成溯祖”。所以其概率表达式应该写作:$P(T_2^{\prime} = i) = (1 - \frac{1}{2N})^i \times \frac{1}{2N}$。从问题的问法 (在$n$次伯努利试验中,试验$k$次才得到第一次成功的概率) 以及该公式,不难看出$T_2^{\prime}$是满足几何分布的。有了这个结论,就知道该几何分布的数学期望为$2N$。这样上面的问题就有了答案:沿着时间向后 (按附图1所示的是向上) 去寻找2条序列的MRCA (即完成溯祖合并),平均需要$2N$代。
问答该问题或许还有另外一个思路。我们知道在上$1$代合并的概率$\frac{1}{2N}$,实际上不管是在当前代,还是中间的任何一代“在上$1$代合并”的概率都为$\frac{1}{2N}$。所以要让合并事件必然发生,平均要等待$2N$代,因为只有$\frac{1}{2N} \times 2N = 1$。
上述推理过程,忽略了一个问题,即从同一亲本来的两个等位基因是不可能同时传递给子代的,所以对于二倍体的等位基因在上一代合并的概率并不是$\frac{1}{2N}$。因此上面的推理更适合于群体大小为$2N$的单倍体群体。但是这里的差异实际上可以忽略不计。
接着上面的讨论。完成溯祖合并平均需要$2N$代,因为$2N$往往很大,所以把原始的等待时间按这个等待时间单位 (每$2N$代为一个单位) 缩小。即令$\frac{T_2^{\prime}}{2N} = T_2$。那么前$i$代不能合并的概率可改写为 $$ P(T_2 > \frac{i}{2N}) = (1 - \frac{1}{2N})^i $$ 当$x$很小时,有$1-x \approx! e^{-x}$。所以 $$ \begin{aligned} P(T_2 > \frac{i}{2N}) &= (1 - \frac{1}{2N})^i \approx! e^{-\frac{i}{2N}} \ P(T_2 \leq! \frac{i}{2N}) &\approx! 1 - e^{-\frac{i}{2N}} \end{aligned} $$ 回想指数分布的概率分布函数 ($P(X \leq! x) = 1 - e^{-x}$) 以及概率密度的公式,可知$T_2$实际上是满足参数$\lambda = 1$的指数分布的随机变量,概率密度为: $$ f(T_2) = e^{T_2} $$ 这里我们该如何理解$T_2$的具体含义呢?$T_2$作为满足指数分布的随机变量,描述的实际上是两次事件发生之间的等待时间,也就是等待下一次溯祖合并所需要的时间。更准确的说,$T_2$是2条随机抽取的序列完成溯祖合并所需要的重标时间 (rescaled time),其均值为$\frac{1}{\lambda} = 1$,方差$\frac{1}{\lambda^2} = 1$。均值$=1$的意义在于,2条随机抽取的序列完成溯祖合并平均需要1个重标时间,而1个重标时间对应$2N$代。
下面我们再引入一个变量$\mu$,用于表示每世代每位点的突变率。群体遗传学上常定义$\mu$为每世代每座位的突变率,这里为了便于处理基因组DNA序列,而采用前一个定义。因为$\mu$ 为“每世代每位点的突变率”,所以令$\mu \times 2N$,将得到经过$2N$个世代后,每个位点上发生的平均突变数。由上面的讨论得知,2条随机抽取的序列完成溯祖合并平均需要$2N$个世代,所以$2N\mu$就相当于2条序列其中之一,到它们的MRCA,每个位点上发生的平均突变数。进一步令$2N\mu \times 2 = 4N\mu$,则为完成溯祖合并的2条序列之间,每个位点上发生的平均突变数。这实际上是一种距离的定义,只是将总突变数按照序列长度平均了。现在令$\theta = 4N \mu$,与上面重标时间的处理类似,我们再让$T_2^{\prime} \times \mu = t_2$,同样会得到$t_2$的概率密度函数,推导过程如下: $$ \begin{aligned} P(T_2^{\prime} > i) &= (1 - \frac{1}{2N})^i \ P(t_2 = T_2^{\prime} \mu > i \mu) &= (1 - \frac{1}{2N})^i \ P(t_2 > i \mu) &= (1 - \frac{1}{\frac{\theta}{2\mu}})^i \ &= (1 - \frac{2\mu}{\theta})^i \approx! (e^{-\frac{2\mu}{\theta}})^i \ &\approx! e^{-\frac{2}{\theta} \cdot! i\mu} \ P(t_2 \leq! i\mu) & \approx! 1 - e^{-\frac{2}{\theta} \cdot! i\mu} \end{aligned} $$ 上式恰为参数$\lambda = \frac{2}{\theta}$的指数分布的概率分布函数,所以得$t_2$的概率密度函数: $$ f(t_2) = \frac{2}{\theta} e^{-\frac{2}{\theta} t_2} $$ 其均值为$\frac{1}{\lambda} = \frac{\theta}{2}$,也就是上面提到的:2条序列其中之一,到它们的MRCA,每个位点上发生的平均突变数。而这里概率密度函数的参数$\lambda$的意义为,当用突变数来衡量等待时间时,发生溯祖合并事件的速率。
以上我们介绍了三种衡量溯祖事件等待时间的方式:
- $T_2^{\prime}$ 以世代数来衡量,均值为$2N$;
- $T_2$ 以每$2N$个世代为单位来衡量,均值为1;
- $t_2$ 以每个位点的突变率来衡量,均值为$\frac{\theta}{2}$。
关于$\theta$,有一个更专门的名词定义,即群体大小参数 (population size parameter)。假设我们随机从一个物种/群体中,在特定基因组位置上,抽取两条DNA序列。那么它们之间的差异的期望值 (均值) 是多少?因为要平均$2N$代之后2条序列才会发生溯祖合并,所以$E(2 \times! T_2^{\prime} \times! \mu) = 4 N \mu = \theta$。因为我们考量的是2条序列之间的距离,所以系数2是必须的。群体大小参数$\theta$实际上是一种群体遗传多样性的度量指标。它反映的是引入遗传变异的突变与降低多态性的遗传漂变之间的平衡。
n条序列的溯祖
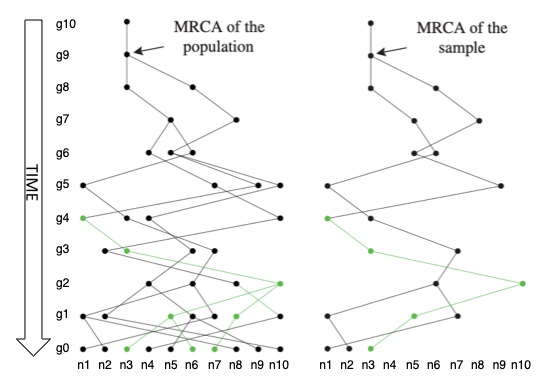
下面我们将2条序列的溯祖,扩展到$n$条序列的溯祖。类似的,先考虑在上一代中$n$条序列中的发生了溯祖事件的概率,也就是说只要$n$序列中的任意一对序列发生了溯祖合并即可。因为任意一对序列在上一代发生溯祖合并的概率为$\frac{1}{2N}$,而$n$序列的两两组合共有${n \choose 2}$个,所以我们要求的概率为${n \choose 2} \frac{1}{2N}$。那么其逆事件“上$1$代中不发生溯祖事件的概率”为:$1-{n \choose 2} \frac{1}{2N}$。和2条序列溯祖的情形一样,接下来考虑:在上$i$代才发生第一次溯祖合并的概率为: $$ P(T_n^{\prime} = i) = \bigg[1-{n \choose 2} \frac{1}{2N}\bigg]^i \times {n \choose 2} \frac{1}{2N} $$ 所以$T_n^{\prime}$表示的是一个样本中$n$条序列等待下一次溯祖事件发生所需要的时间,同样满足几何分布,均值为$\frac{2N}{{n \choose 2}}$。这是考虑所有$n$条序列的情形,那么只考虑序列样本中的一部分,比如$j$条序列呢?只需要形式上将$n$改为$j$即可。这种形式上的改动看似简单,但其实这里隐含着溯祖模型的一个重要特点,也是它的优点,即群体中与抽取序列不相关的样本可以完全忽视 (附图2)。
令$T_j = T_j^{\prime} / (2N)$,即用$2N$代重标时间,有: $$ \begin{aligned} P(T_j^{\prime} > i) &= \bigg[ 1 - {j \choose 2}\frac{1}{2N} \bigg]^i \ P(T_j > \frac{i}{2N}) &= (1 - \frac{{j \choose 2}}{2N})^i \ &\approx! ( e ^ {-\frac{{j \choose 2}}{2N}})^i \ &\approx! e^{-{j \choose 2} \frac{i}{2N}} \ &\approx! e^{- \frac{j(j-1)}{2} \frac{i}{2N}} \ P(T_j \leq! \frac{i}{2N}) &\approx! 1 - e^{- \frac{j(j-1)}{2} \frac{i}{2N}} \end{aligned} $$ 所以$T_j$的概率密度为 $$ f(T_j) = \frac{j(j-1)}{2} e ^{-\frac{j(j-1)}{2} T_j} $$ 均值为$\frac{2}{j(j-1)}$,方差为$[\frac{2}{j(j-1)}]^2$。
令$t_j = T_j^{\prime} \mu$,即用每个位点的突变率重标时间,有: $$ \begin{aligned} P(T_j^{\prime} > i) & = \bigg[ 1 - {j \choose 2}\frac{1}{2N} \bigg]^i \ P(T_j^{\prime} \mu > i \mu) & = \bigg[ 1 - {j \choose 2}\frac{1}{2N} \bigg]^i \ P(t_j > i \mu) & = \bigg[ 1 - \frac{j(j-1)}{2} \frac{1}{2N} \bigg]^i \ & \approx! (e^{-\frac{j(j-1)}{4N}})^i \ & \approx! (e^{-\frac{j(j-1)\mu}{\theta}})^i \ & \approx! e^{-\frac{j(j-1)}{\theta} \cdot! i\mu} \ P(t_j \leq! i \mu) & \approx! 1 - e^{-\frac{j(j-1)}{\theta} \cdot! i\mu} \ \end{aligned} $$ 所以$t_j$的概率密度为 $$ f(t_j) = \frac{j(j-1)}{\theta} e^{- \frac{j(j-1)}{\theta} t_j} $$ 均值为$\frac{\theta}{j(j-1)}$,方差为$[\frac{\theta}{j(j-1)}]^2$。
需要注意的是,$t_j$的概率描述的是$j$条序列完成一次溯祖合并变为$j-1$条序列的概率。而$j$条序列的两两组合共有${j \choose 2} = \frac{j(j-1)}{2}$种可能。所以完成溯祖的那两条序列,出现的概率为$\frac{2}{j(j-1)}$。也就是说当给定一个基因树$G$时 (如图2右图蓝线所示的基因树),每次所能观察到的溯祖事件,其等待时间的概率为 $$ \frac{2}{j(j-1)} \times \frac{j(j-1)}{\theta} e^{- \frac{j(j-1)}{\theta} t_j} = \frac{2}{\theta} e^{- \frac{j(j-1)}{\theta} t_j} $$
几点性质
树的高度
树的高度,即从树末端到根部的距离,也就是在MRCA溯祖合并的等待时间$T_{MRCA}$。对于任意一个“标记历史”,从当前代到MRCA,共有$n-1$个溯祖合并时间$T_n, T_{n-1}, ... , T_2$。其中$T_n$是在上一代溯祖的等待时间,$T_2$是从距根部最近的一代 (只有2个样本) 在MRCA完成溯祖的等待时间。所以树的高度 $$ T_{MRCA} = T_n + T_{n-1} + ... + T_2 $$ 这是一个$n-1$个独立指数等待时间的求和问题。 $$ \begin{aligned} E(T_{MRCA}) &= E(T_n + T_{n-1} + ... + T_2) = \sum_{j=2}^n E(T_n) = \sum_{j=2}^n \frac{2}{j(j-1)} = 2 \sum_{j=2}^n(\frac{1}{j-1} - \frac{1}{j}) = 2(1- \frac{1}{n}) \ V(T_{MRCA}) &= \sum_{j=2}^n V(T_j) = \sum_{j=2}^n [\frac{2}{j(j-1)}]^2 = 8\sum_{j-1}^{n-1} \frac{1}{j^2} - 4(3-\frac{2}{n}-\frac{1}{n^2}) \end{aligned} $$
注意, 当$n$较大时,$E(T_{MRCA}) \approx 2$ (以每$2N$个世代为单位来衡量),我们已经知道$T_2 = 1$。由此可知,所有样本 (序列,或世系) 完成溯祖合并所用的时间,仅仅是最后两个样本合并所需时间的2倍。此外,同样当$n$较大时,$V(T_{MRCA}) \approx \frac{8 \pi^2}{6} - 12 \approx 1.16$,而$V(T_2) = 1$,所以$T_{MRCA}$的方差也主要是由$T_2$的方差贡献的。
树的长度
树的长度由系谱树上各分枝的长度之和构成,即$T_{total} = \sum_{j=2}^{n} jT_j$。有均值和方差如下 $$ \begin{aligned} E(T_{total}) &= E\bigg(\sum_{j=2}^n jT_j \bigg) = \sum_{j=2}^n \frac{2}{j-1} = 2\sum_{j=1}^{n-1} \frac{1}{j}\ V(T_{total}) &= \sum_{j=2}^n j^2 V(T_j) = \sum_{j=2}^n j^2 (\frac{2}{j(j-1)})^2 = 4 \sum_{j=1}^{n-1} \frac{1}{j^2} \end{aligned} $$ 当$n$较大时,$E(T_{total}) \approx! 2(\gamma + \log! n)$,其中$\gamma = \lim_{n \to \infty} \sum_{j=1}^n \frac{1}{j} - \log(n) \approx 0.577$为欧拉常数。方差$V(T_{total}) \approx \frac{2\pi^2}{3} \approx 6.579$。所以树的长度随$n$的增加,增长的非常缓慢,因为主要的增加的长度主要分布在树末端较短的细枝上。
样本包含根的概率
对于$n$个样本的群体的MRCA,也就是整个群体的MRCA,其出现的概率为$\frac{n-1}{n+1}$。也就是说对于$n=2,3,9$,MRCA的概率分别为$1/3, 1/2, 0.8$。也就是说即使是抽取较少的样本,都有较大的可能包含整个群体的根。