
进化的概率模型简介
概述——进化模型的重要性
由于高通量测序、功能基因组学和计算技术的进步,过去的十年见证了进化生物学的一场革命。基因组革命为用合理的成本从非模式生物中快速生成大规模测序数据开辟了可能性。然而,尽管已经有新的方法来处理和组装这些数据,但将这些数据转化为有意义的生物学知识的方法论进展仍然落后。事实上,未来十年的主要挑战之一仍然是解开基因组变化与自然界中表型多样性之间的联系,并进一步破译导致这种多样性的进化力量。这样的研究努力将导致一个更具解释性的进化论,对生命科学的所有分支都有影响,从农业到生态学,再到医学。发现这些联系是一项困难的任务,需要新的生物学启发的 (biologically-inspired) 计算方法,以识别被选择的候选位点,并将这些位点与表型差异关联起来。在后基因组时代,分子进化的概率模型是数据分析动力源。这些模型构成了各种生物信息学应用的支柱,如系统发育重建和功能位点预测。这些模型是一个试图描绘进化随机性质的尝试,并将其建立在一个健壮的统计推断框架内。构建进化模型使我们能够从数学上描述各种生物现象,估计相关参数,如自然选择的强度和进化事件的速率,并测试不同的假设,如确定几种替代进化路径中哪一种更有可能。设计模型的一个主要挑战是在不过度参数化的情况下,捕获所要研究的生物学过程的关键元素。相关模型虽然多数时候是不够充分的,但在某些关键问题的解决上是有力的。在分子系统发育学中,马尔可夫模型是一类特殊的随机模型,广泛应用于分析序列数据,和量化基因和基因组的进化动力学。
离散时间马尔可夫链
使用计算机模拟进化过程,将有助于思考进化的概率模型。具体地说,我们感兴趣的是描述序列如何随时间而变化,以及哪些参数控制了这个进化过程。简单起见,下面我们只描述一个序列位点的演化;假设所有位点都独立演化,这个过程可以重复$N$次,生成一个长度$N$的序列 (此序列是在一个位点上沿着时间发生的位点组成变化序列,而不是像DNA分子那样空间上线性排列的序列)。我们首先绘制在最初一代这个位点上的状态,即祖先 (ancestor)。假设所有核苷酸的概率都是相等的,这相当于掷一个有$4$种可能结果的骰子 ($A$、$C$、$G$或$T$)。接下来,我们想决定这个位点在下一代的状态。它可能变异,也可能不变异。如果它发生变异,它就会以相等的概率变成其它任一不相同的核苷酸。我们可以将这个模型写成矩阵形式: $$ M_{ij} = \left( \begin{matrix} 1-3\epsilon & \epsilon & \epsilon & \epsilon\ \epsilon & 1-3\epsilon & \epsilon & \epsilon \ \epsilon & \epsilon & 1-3\epsilon & \epsilon \ \epsilon & \epsilon & \epsilon & 1-3\epsilon \end{matrix} \right) $$ 该矩阵表示一个具有$4$个状态的简单离散时间马尔可夫模型 (discrete-time Markov model)。该模型是离散的,因为$t$,对应于代数 (the number of generations) 时间,只能取离散值 $(0, 1, 2, ...)$。$M_{ij}$是从核苷酸$i$开始,经过一代后以核苷酸$j$结束的概率。因为每行都包含所有可能的事件,所以每行的和必须等于$1$。在该模型中有一个自由参数 (free parameter),$0 \leq \epsilon \leq 1/3$,它决定了进化的速率。如果我们运行多代模拟,与较大的$\epsilon$相比,低$\epsilon$值的突变预期将更少。事实上,每一代都有两种可能性:要么是发生了突变,概率为$3\epsilon$,要么是位点保持不变,概率为$1 - 3\epsilon$。因此,在单一代上突变数的期望为$3\epsilon$。
假设现在使用这个模型来模拟1000代的演化过程,我们感兴趣的是计算得到在第$1000$代是核苷酸$C$的概率,假定第$0$代从核苷酸$A$开始。一种幼稚的方法是根据矩阵$M$模拟$1000$代的进化过程,并记录最后的核苷酸 (注意,我们总是在第$0$代用$A$开始模拟)。这代表了一个可能的进化结果。为了估计所想要的概率,我们多次重复这个模拟过程 (比如100万次)。“以$C$结束”的频率就是目标概率的一个很好的估计。
然而,我们可以利用马尔可夫链理论来解析地 (analytically) 计算这个概率。首先计算 (而不是模拟) 事件从$A$开始,两代后以$C$结束后的概率,我们用$P_{A,C}(2)$表示这个概率。此事件的执行过程可以描述为:“$A$在一代后将变为某未知状态$X$” ($P_{A,X}(1)$),然后$X$再变为$C$ ($P_{X,C}(1)$)。根据全概率公式,我们可以通过总结所有可能值来表示这个概率: $$ P_{A,C}(2) = P_{A,A}(1)P_{A,C}(1) + P_{A,C}(1)P_{C,C}(1) + P_{A,G}(1)P_{G,C}(1) + P_{A,T}(1)P_{T,C}(1) $$ 我们注意到,这里有一个隐式的假设:从第0代$A$到第$1$代$X$的概率,与第1代$A$到第2代$X$的概率相同。更正式地说,我们假设马尔可夫链是时间同质的 (time homogenous)。在概率术语中,用$X(k)$表示$k$代后的核苷酸;矩阵$M$表示任意两代间的转移概率 (transition probabilities),使用这个符号,我们可以将$M$表示为 $$ M_{ij} = P(X(k+1) = j | X(k) = i) = P_{i,j}(1) $$ 请注意,这种等式直接源于时间同质性的假设。在矩阵形式中,我们将把$M$视为$P(1)$,即马尔可夫矩阵表示一代后发生变化的概率。这里,我们还隐式地假设:从第$1000$代的$A$到第$1001$代的转移概率仅由$M$决定,而不依赖于第$0$到$1000$代事件发生的历史。这是马尔可夫过程的另一个重要属性,被称为无记忆性 (memorylessness)。
返回到等式2,我们可以注意到这个计算与$M$中的$A$行 (第一行) 和$C$列 (第二列) 的点积 (dot product) 相同。让我们用$P(2)$表示两代后每对核苷酸之间的转移概率矩阵 (一般的,$P(k)$表示$k$代后的转移概率矩阵)。每个元素的计算采用与等式2相同的方式执行: $$ P(2) = \left( \begin{matrix} P_{AA}(2) & P_{AC}(2) & P_{AG}(2) & P_{AT}(2) \ P_{CA}(2) & P_{CC}(2) & P_{CG}(2) & P_{CT}(2) \ P_{GA}(2) & P_{GC}(2) & P_{GG}(2) & P_{GT}(2) \ P_{TA}(2) & P_{TC}(2) & P_{TG}(2) & P_{TT}(2) \end{matrix} \right) $$ 基于以上考虑可知,$P(2) = P(1)^2 = M^2$,更一般的形式有 $P(k) = P(1)^k$和$P(n+m) = P(n)P(m)$。最后这一等式的重要性将在下文中变得更加清楚。因此,在一个离散的时间过程中,通过了解矩阵$M$,可以推导出任意$k$代后的转移概率$P(k)$。因此,$M=P(1)$有时被称为离散时间马尔可夫链的生成器矩阵 (generator matrix)。把所有这些都放在一起,我们得到了: $$ P_{ij}(k) = P(X(k) = j | X(0) = i) = [M^k]_{i,j} $$
连续时间马尔可夫链
上述离散时间马尔可夫链的一个自然推广是用一个度量时间的连续参数$t$来代替离散的代数时间。实际上,这样一个$t$可以在区间$[0, \infty)$上有任何取值的过程被称为连续时间马尔可夫链 (continuous time Markov chain)。与离散过程类似,$X(t)$表示特定位点在时间$t$时的状态。在这里,我们不仅要计算$P(1)$和$P(2)$,还要计算$P(2.335)$。为了理解如何计算这样的矩阵,我们记得对于离散马尔可夫过程,有$P(t_1+t_2)=P(t_1)P(t_2)=P(t_2)P(t_1)$,这对于连续时间马尔可夫链也是成立的。由此可见,我们可以得到: $$ P(t_1 + t_2) - P(t_1) = P(t_1)P(t_2) - P(t_1) = P(t_1)(P(t_2) - I) $$ 其中$I$为单位矩阵 (identity matrix)。注意,$P(0) = I$,因为$P_{ii}(0) = P(X(0) = i |X(0)=i) = 1$,类似的有$P_{ij}(0) = 0$对于所有$i \neq j$。上式意味着对于任意$t_2 \neq 0$: $$ \frac{P(t_1 + t_2) - P(t_1)}{t_2} = \frac{P(t_1)(P(t_2) - I)}{t_2} $$ 现在用$t$替换$t_1$,$\Delta! t$替换$t_2$,然后当$\Delta! t$趋近于$0$时,得到: $$ \lim_{\Delta! t \to 0} \frac{P(t + \Delta! t) - P(t)}{\Delta! t} = \lim_{\Delta! t \to 0} \frac{P(t)(P(\Delta! t) - P(0))}{\Delta! t} = \lim_{\Delta! t \to 0} P(t)\frac{P(\Delta! t) - P(0)}{\Delta! t} = P(t)P^{\prime}(0) $$ 其中最后一个等式源于时间$0$处导数的定义,而方程的左侧是时间$t$处的导数的定义,因此: $$ P^{\prime}(t) = P(t)P^{\prime}(0) $$ 在上述方程中,我们使用了矩阵导数的概念:如果对于矩阵$P(t)$,矩阵的每个元素都是$t$的函数,那么在$P^{\prime}(t)$中,每个元素都是对应的$P(t)$项的导数。$P^{\prime}(0)$是一种固定矩阵,称为瞬时速率矩阵 (instantaneous rate matrix),通常表示为$Q \equiv! P^{\prime}(0)$。
当给定$Q$时,我们可以计算任意时间间隔$t$的转移概率矩阵$P(t)$,因此$Q$通常被称为连续马尔可夫过程的生成矩阵。$P(t)$中每行的元素总和为$1$: $$ P_{i,1}(t) + P_{i,2}(t) + P_{i,3}(t) + ... + P_{i,n}(t) = 1 $$ 其中,$n$是可能状态的数目 (核苷酸为4个)。对等式10两边求导,我们得到: $$ P_{i,1}^{\prime}(t) + P_{i,2}^{\prime}(t) + P_{i,3}^{\prime}(t) + ... + P_{i,n}^{\prime}(t) = 0 $$ 特别地,当$t=0$时,有 $$ P_{i,1}^{\prime}(0) + P_{i,2}^{\prime}(0) + P_{i,3}^{\prime}(0) + ... + P_{i,n}^{\prime}(0) = 0 $$ 因为$Q = P^{\prime}(0)$, 又有 $$ Q_{i,1} + Q_{i,2} + Q_{i,3} + ... + Q_{i,n} = 0 $$
因此,$Q$每行中的元素之和为零。对于$i \neq j$,$P_{i,j}(t)$是一个增函数,当$t=0$时为零,当$t>0$时是非负的。因此,每个非对角元素$Q_{i,j}=P_{i,j}^{\prime}(0)$都是非负的,表示从状态$i$到状态$j$的瞬时转移速率,而每个对角元素$Q_{i,i}$都为负,等于该行中所有其他元素求和后取负。$Q_{i,i}$表示远离状态$i$的总瞬时转移速率。
上面的等式9实际上是一个矩阵形式的微分方程: $$ \frac{\mathrm{d} P(t)}{\mathrm{d}t} = P(t)Q $$ 这个方程在边界条件$P(0) = I$的解为: $$ P(t) = e^{Qt} = I + Qt + \frac{(Qt)^2}{2!} + \frac{(Qt)^3}{3!} + ... $$ 我们将注意到,这个幂级数总是收敛的。计算矩阵指数是数值分析中一个被充分研究的课题,这里不讨论。然而,对于某些类型的简单模型,可以直接得到$P(t)$的封闭解。下面我们推导出最简单的核苷酸模型的$P(t)$矩阵,Jukers & Cantor (JC) 模型。
Jukers & Cantor模型
关于核苷酸的最简单连续时间马尔可夫模型,假设每两个不同的核苷酸之间的转移概率是相同的。用$f(t)$表示,在矩阵形式中,$P(t)$则为: $$ P(t) = \left( \begin{matrix} 1-3f(t) & f(t) & f(t) & f(t) \ f(t) & 1-3f(t) & f(t) & f(t) \ f(t) & f(t) & 1-3f(t) & f(t) \ f(t) & f(t) & f(t) & 1-3f(t) \end{matrix} \right) $$ 根据$Q$的定义,我们可以得到: $$ Q = P^{\prime}(0) = \left( \begin{matrix} -3f^{\prime}(0) & f^{\prime}(0) & f^{\prime}(0) & f^{\prime}(0) \ f^{\prime}(0) & -3f^{\prime}(0) & f^{\prime}(0) & f^{\prime}(0) \ f^{\prime}(0) & f^{\prime}(0) & -3f^{\prime}(0) & f^{\prime}(0) \ f^{\prime}(0) & f^{\prime}(0) & f^{\prime}(0) & -3f^{\prime}(0) \end{matrix} \right) $$ 如果用$\alpha$表示$f^{\prime}(0)$,即: $$ Q = \left( \begin{matrix} -3\alpha & \alpha & \alpha & \alpha \ \alpha & -3\alpha & \alpha & \alpha \ \alpha & \alpha & -3\alpha & \alpha \ \alpha & \alpha & \alpha & -3\alpha \end{matrix} \right) $$ 由这个矩阵产生的随机过程(假设所有核苷酸的频率相同) 被称为Jukers & Cantor (JC) 模型 (Jukers and Cantor, 1969)。应用等式14到JC模型中,我们得到: $$ \frac{\mathrm{d}}{\mathrm{d}t} \left( \begin{matrix} 1-3f(t) & f(t) & f(t) & f(t) \ f(t) & 1-3f(t) & f(t) & f(t) \ f(t) & f(t) & 1-3f(t) & f(t) \ f(t) & f(t) & f(t) & 1-3f(t) \end{matrix} \right) = \ \left( \begin{matrix} 1-3f(t) & f(t) & f(t) & f(t) \ f(t) & 1-3f(t) & f(t) & f(t) \ f(t) & f(t) & 1-3f(t) & f(t) \ f(t) & f(t) & f(t) & 1-3f(t) \end{matrix} \right) \left( \begin{matrix} -3\alpha & \alpha & \alpha & \alpha \ \alpha & -3\alpha & \alpha & \alpha \ \alpha & \alpha & -3\alpha & \alpha \ \alpha & \alpha & \alpha & -3\alpha \end{matrix} \right) $$ 如果我们只关注此方程左侧第一行和第二列中的元素 (并将所有其他元素标记为“*”),并使用矩阵乘法的定义: $$ \left( \begin{matrix} * & f^{\prime}(t) & * & * \ * & * & * & * \ * & * & * & * \ * & * & * & * \end{matrix} \right) = \left( \begin{matrix} 1-3f(t) & f(t) & f(t) & f(t) \ * & * & * & * \ * & * & * & * \ * & * & * & * \end{matrix} \right) \left( \begin{matrix} * & \alpha & * & * \ * & -3\alpha & * & * \ * & \alpha & * & * \ * & \alpha & * & * \end{matrix} \right) $$
也就是 $$ f^{\prime}(t) = [1 - 3f(t)]\alpha + f(t)(-3\alpha) + f(t)\alpha + f(t)\alpha $$ 经过简化后,可得到以下正则微分方程: $$ \frac{\mathrm{d}f(t)}{\mathrm{d}t} = \alpha(1-4f(t)) $$ 此方程式可以非正式地写为: $$ \frac{\mathrm{d}f(t)}{1-4f(t)} = \alpha 4 \mathrm{d}t $$ 两边积分,可得: $$ \frac{ln(1 - 4f(t))}{-4} = \alpha! t + C $$ 然后得: $$ f(t) = \frac{1}{4} - \frac{e^{-4\alpha! t - 4C}}{4} $$ 实际上$f(t)$即$P_{i,j}(t)$,当$i \neq j$,又因为$P(0) = I$,所以$f(0)=0$,可得$C = 0$,所以 $$ f(t) = \frac{1}{4} - \frac{e^{-4\alpha! t}}{4} $$ 相似的,因为$1-3f(t)$即$P_{i,i}(t)$,所以可得: $$ P_{i,j}(t) = \begin{cases} \frac{1}{4} - \frac{e^{-4\alpha! t}}{4} & \text{if}\: i \neq j,\ \frac{1}{4} + \frac{3e^{-4\alpha! t}}{4} & \text{if}\: i=j\ \end{cases} $$
似然方程与矩阵标准化
在JC模型中,给定参数 $\alpha$和$t$的值,我们可以模拟一条初始序列和并让它在计算机里演化,也可以为进化场景编写显式的概率表达。从一条只有一个位点的序列开始,假设状态为$A$。根据JC模型,从$A$开始的概率是$P(A)=0.25$,因为JC模型假定所有核苷酸的概率是相等的。这意味着在所有模拟中,从一个位点开始,平均有$1/4$的机会将从字符$A$开始。现在,考虑在时间$t=0.5$之后,该位点上的状态由$A$变成了$C$。我们用下图来表示此场景:
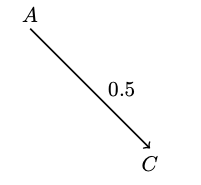
在JC模型下模拟这种场景的概率为$P(A) \cdot! P_{A,C}(0.5)$。如果我们将这两条序列 (实际上也是两个位点) 作为我们的数据,以$D$表示,并用$M$表示模型,我们可以用$P(D|M)$来表示这个概率,即当给定模型及其参数时,得到数据的概率。这被称为模型的似然 (the likelihood of the model)。现在考虑一下更复杂的情况,序列$ACGGT$进化到$ACGAC$,同样是经历时间$t=0.5$。模型的似然是数据中每个位置的概率的乘积: $$ (P(A) \cdot! P_{A,A}(0.5)) \cdot! (P(C) \cdot! P_{C,C}(0.5)) \cdot! (P(G) \cdot! P_{G,G}(0.5)) \cdot! (P(G) \cdot! P_{G,A}(0.5)) \cdot! (P(T) \cdot! P_{T,C}(0.5)) $$ 请注意,概率的乘积反映了这样一个假设,即给定模型参数、树及其分枝,不同位点彼此独立演化。这个假设显然是生物学现实的过度简化,但它包含在分子进化中使用的绝大多数模型中,因为它允许快速计算似然函数。在JC模型下,似然同时取决于$t$和$\alpha$ (每个$P_{i,j}(t)$项都取决于$t$和$\alpha$)。因此,我们可以称之为$L(t, \alpha)$。
在审视生物序列时,我们不知道产生它们的参数值。在许多统计应用中很常见,推断这些值通常涉及到寻找似然函数最大化的参数值。这些值被称为最大似然估计 (maximum likelihood estimates, MLE)。但是,请注意,有无限多个$(t, \alpha)$对可以使似然函数最大化。这是因为在所有这些表达式中,$t$和$\alpha$总是以一个整体出现,$\alpha! t$。因此,我们总是可以将$\alpha$乘以一个常数值,并将$t$除以那个常数值,其似然值将保持不变。这是统计学中一个众所周知的现象,这种模型被称为不可识别参数的 (non-identifiable parametric),即在似然函数和一组模型参数值之间没有一对一的映射关系。请注意,此不可识别性并不限于$JC$模型;我们看到等式15,$P(t)$取决于$Qt$。因此,我们总是可以将$Q$乘以一个因子,并将$t$除以相同的因子,而$P(t)$函数 (因此似然函数) 将保持不变。然而,尽管有无限数量的MLE $(t,\alpha)$对,我们还是可以唯一地推断出它们的乘积$\alpha! t$。对于上述示例,并使用$JC$模型,$\alpha! t=0.19$使似然函数最大化。
我们现在将看到,当序列演化$t$个单位时间后,乘积$\alpha! t$也与平均替代数有关。
对于演化的连续时间马尔可夫过程,$d$为沿长度为$t$的分枝的期望替代数,可以用以下表达式来计算: $$ d = - t \sum_i P(i) \cdot! Q_{i,i} $$ 我们在这里不展示这个公式的推导。对于上述的$JC$模型,这将得到$d=3 \alpha! t$。这表明我们可以选择任何一个使似然函数最大化的$(t, \alpha)$对,基于这对参数计算$d$,并获得与MLEs相关分枝上的期望替代数。或者,我们可以设置$\alpha = 1/3$,这将意味着$d=t$。由此得到的JC模型是: $$ P_{i,j}(t) = \begin{cases} \frac{1}{4} - \frac{e^{-4 t}}{4} & \text{if}\: i \neq j,\ \frac{1}{4} + \frac{3e^{-4 t}}{4} & \text{if}\: i=j\ \end{cases} $$ 根据等式29,这可以推广到JC模型之外的模型,$d$将等于$t$,如果 $$ \sum_i P(i) \cdot! Q_{i,i} = -1 $$ 我们总是可以将$Q$矩阵乘以一个固定的因子,从而使等式31成立。这被称为矩阵标准化 (matrix normalization),遵循这个过程,$t$的估计实际上对应于长度$t$分分枝的期望替代数,而不是时间。
可逆性、同质性和平稳性
马尔可夫过程的时间可逆性可用以下等式表达: $$ P(i) \cdot! P_{i,j}(t) = P(j) \cdot! P_{j,i}(t) $$ 也就是说,以字符$A$开始并沿着长度为$t$的分枝演化并以字符$C$结束字符的概率,与以字符$C$开始沿着相同的分枝演化并以字符$A$结束的概率相同。更一般地说,这表明当我们有两个序列,假设第一个是祖先,第二个是后代,获得两条序列的概率与“假设第一个是后代和第二个是祖先”是相同的。更一般地,我们可以沿分枝$t$的任何地方选择“最古老” (祖先根) 点,该点将分枝$t$分为两个分段$t_1$和$t_2=t - t_1$,分段将两条序列连接到新的根。可逆性强制地让 (对于不同的根位置) 似然函数保持不变,也就是说,无论我们如何选择$t_1$,它都将保持不变。我们在这里展示了两个单个字符序列的非常简单的情况:
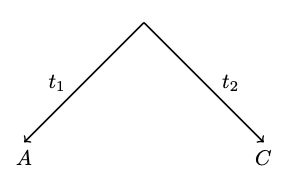
序列之间的分割定义了最简单的分叉树。沿树的似然计算通过根的所有可能状态$x$给出总和: $$ \sum_{x}P(x) \cdot! P_{x,A}(t_1) \cdot! P_{x,C}(t_2) $$ 利用前两个项的可逆性条件,我们得到了: $$ \sum_x P(A) \cdot! P_{A,x}(t_1) \cdot! P_{x,C}(t_2) $$ 可以将第一项从求和表达式中移出: $$ P(A) \sum_x P_{A,x}(t_1) \cdot! P_{x,C}(t_2) $$ 利用马尔可夫性质,我们可以将该方程简化为: $$ P(A)P_{A,C}(t_1 + t_2) $$ 因此我们看到,似然值取决于两段分枝的总长度,表明只要总长度保持固定,我们就可以移动根的位置。甚至我们可以将一个分枝设置为零,而将另一个分枝设置为$t_1+t_2$,其似然值将保持不变,即一个序列可以被认为是另一个序列的祖先:
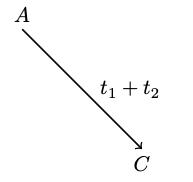
对于有三个类群的无根树,可以将根放在三个分枝的任何一点。同样的论点表明,所有的根位置对两个序列的结果都有相同的似然,也适用于这种情况,事实上,可逆性条件使所有从同一无根树派生出来的有根树都有同样的似然。这表明,当假设一个可逆的模型时,就不可能推断出树根的位置。然而,可逆性允许非常有效的算法来推断分枝的长度,以及搜索最高似然的树,通过使用所谓的滑轮原理。
如果我们观察一个离散的 (或连续的) 时间马尔可夫链,通常会对该链运行很长一段时间后处于特定状态的概率感兴趣。在某些情况下,我们发现当$t$接近无穷时,$P(t)$收敛于一个特定形式,其中所有行都是相同的,因此$P(\infty!)=\pi! \cdot! I$是$\pi$大小为$n$的行向量,其中$\pi_i$是无限时间后到达状态$i$的概率。此外,概率向量$\pi$对于转移矩阵$P(1)$是不变的,即$\pi !\cdot! P(1)=\pi$。这种情况被称为平稳链 (stationary chains),而$\pi$是唯一的平稳概率向量。一旦链达到平稳状态,它就会保持下去。例如,JC模型对于每个字符的平稳概率正好为$0.25$。
同质性 (homogeneity) 意味着一个单一的$Q$矩阵描述了整个进化过程。相比之下,分枝异构模型假设树的不同分枝具有不同的$Q$矩阵。
目前,绝大多数的系统发育研究假设的模型是可逆的、平稳的和同质的。然而,很明显,这种模型在许多情况下是对现实来说过于简化。例如,细菌基因组在同源区域的GC成分上有很大的差异,这明显违反了这些假设。
核苷酸替代模型
如上所述,在JC模型中,用最简单的核苷酸转移概率表示 (见等式27),假设所有不同核苷酸之间的瞬时替代速率是相同的,这意味着平稳频率等于$0.25$。然而,有充足的实证表明,核苷酸之间的替代概率是有所不同的。在过去的50年里,这个基本的建模方案得到了大量的扩展,每一次有关于核苷酸进化模式的不同假设。其中,Kimura-two-parameters模型 (K2P, Kimura, 1980) 是模型扩展迈出的第一步,它缓解了关于——”转换(两对嘌呤之间或两对嘧啶之间的替代) 和颠换 (嘌呤和嘌啶之间的替代) 的概率是相等的“,这一假设。K2P模型的$Q$矩阵为: $$ Q[K2P] = \left( \begin{matrix} -\alpha - 2\beta & \beta & \alpha & \beta \ \beta & -\alpha - 2\beta & \beta & \alpha \ \alpha & \beta & -\alpha - 2\beta & \beta \ \beta & \alpha & \beta & -\alpha - 2\beta \end{matrix} \right) $$
其中$\alpha$是转换的速率,$\beta$是颠换的速率。
矩阵标准化对矩阵引入了一个约束,即给定一个特定的$\alpha$值,就可以计算$\beta$值。事实上,在标准化之后,可以只使用一个参数来重写K2P矩阵,即转换-颠换速率比 (transition-transversion rate ratio)。可能的转换-颠换速率比有无限多个,应该使用哪一个呢?按照最大似然法原则,应该用使似然函数最大化的那个。该“转换-颠换速率比”为自由参数,因为它是使用基于数据的似然值来估计的。虽然其他类型的参数化也是可能的,但每一种都捕获生物学过程的不同侧面。在替代率方面最复杂的模型,假设替代率是对称的 (即对于所有的$i$和$j$有$Q_{i,j}=Q_{j,i}$) ,可用一个具有6个参数 (五个自由参数) 的矩阵描述,并表示为SYM模型:
$$
Q[SYM] = \left( \begin{matrix} -\alpha - \beta - \gamma & \beta & \alpha & \gamma \
\beta & -\beta - \delta - \epsilon & \delta & \epsilon \
\alpha & \delta & -\alpha - \delta - \eta & \eta \
\gamma & \epsilon & \eta & -\gamma - \epsilon - \eta \end{matrix} \right)
$$
由于JC、K2P和SYM矩阵是对称的 (对于所有$i$和$j$有$Q_{i,j}=Q_{j,i}$),因此所有核苷酸的平稳频率都等于$0.25$。如上所述,生物数据集通常具有偏倚的核苷酸频率。对这类模型的一个重要扩展依赖于非对称矩阵,它可以导致任何期望的平稳核苷酸频率,由$\pi$表示。为此,在模型中添加了三个额外的自由参数:$\pi_A$、$\pi_C$和$\pi_G$,表示$A$、$C$和$G$的频率 ($T$的频率$\pi_T$是非自由的,受到所有频率的和为$1$的限制)。将这些参数合并到JC、K2P和SYM模型中,就产生了一组扩展的模型,分别称为F81、HKY和一般时间可逆模型 (General Time Reversible model, GTR),一般来说,包含这些平稳频率的模型通常用+F表示。为了保证时间可逆性,此类模型采用以下一般形式:
$$
Q[GTR] = \left( \begin{matrix} -... & \beta \pi_C& \alpha \pi_G & \gamma \pi_T \
\beta \pi_A & -... & \delta \pi_G & \epsilon \pi_T \
\alpha \pi_A & \delta \pi_C & -... & \eta \pi_T \
\gamma \pi_A & \epsilon \pi_C & \eta \pi_G & -... \end{matrix} \right)
$$
注意,这个模型仍然是时间可逆的,因为它满足$\pi_i Q_{i,j}=\pi_jQ_{j,i}$,对于所有$i$和$j$。
密码子替代模型
上面模型都假设可能的状态是$4$个DNA核苷酸,即,我们使用一个只有$4$个字母的字母表。当分析蛋白质编码的核苷酸序列时,转移速率受到它们所编码蛋白质的高度影响。在这种情况下,密码子$I$和$J$之间的转移速率可能取决于核苷酸替代的类型、氨基酸替代的类型以及密码子的使用 (即目标密码子的频率)。密码子模型最初是由Goldman和Yang (1994)、Muse和Gaut (1994) 同时开发的。该模型通常由一个61×61密码子转移速率矩阵表示,它描述了从密码子$I=i_1i_2i_3$到密码子$J=j_1j_2j_3$的瞬时替代率。该矩阵根据密码子中三个子位点的类型和对编码氨基酸的影响,对核苷酸替代分配不同的速率: $$ Q_{I,J} = \begin{cases} \alpha_{i_k j_k} \pi_J & \text{I\,and\,J\,differ\,by\,one\,synonymous\,substitution\,at\,codon\,site\,}k \ r(A_I, A_J) \alpha_{i_k j_k} \pi_J & \text{I\,and\,J\,differ\,by\,one\,nonsynonymous\,substitution\,at\,codon\,site\,}k\ 0 & \text{I\,and\,J\,differ\,by\,more\,than\,one\,nucleotide} \end{cases} $$ 这里,$\pi_J$是密码子$J$的频率,$\alpha_{i_k j_k}$是核苷酸$i_k$到核苷酸$j_k$之间的替代速率,可以根据任何时间可逆的核苷酸替代模型进行参数化,如GTR或HKY。$r(A_I, A_J)$是由密码子$I$和$J$分别编码的氨基酸$A_I$和$A_J$之间的替代率。请注意,为了保持可逆性特性,$r(A_I, A_J)$应等于$r(A_J, A_I)$。在Goldman和Yang最初建立的模型中,$r(A_I, A_J)$依赖于两个因素: (1) 氨基酸$A_I$和$A_J$之间的物理化学距离; (2) 针对作用于编码蛋白质的选择强度的自由参数。在Muse和Gaut模型中,$r(A_J, A_I)$仅用选择强度参数ω表示,它指定了同义-非同义替代率之比: $$ Q_{I,J} = \begin{cases} \alpha_{i_k j_k} \pi_J & \text{I\,and\,J\,differ\,by\,one\,synonymous\,substitution\,at\,codon\,site\,}k \ \omega \alpha_{i_k j_k} \pi_J & \text{I\,and\,J\,differ\,by\,one\,nonsynonymous\,substitution\,at\,codon\,site\,}k\ 0 & \text{I\,and\,J\,differ\,by\,more\,than\,one\,nucleotide} \end{cases} $$
氨基酸替代模型
在氨基酸水平上分析序列需要一个$20 \times 20$维的瞬时速率矩阵。如果每对氨基酸之间的替代率被定义为一个自由参数,需要根据数据估计$190$个参数 (矩阵标准化后是189个),这既需要计算资源的支持,也需要大量的数据来提高推断的准确度。因此,对于蛋白质序列,通常使用一个预先计算的矩阵,其参数是基于一个非常大的蛋白质数据集推断出来的。例如,Dayhoff et al. (1978) 整理修正了当时所有可用蛋白质序列的一个图集,并基于类最大简约的程序估计了替代率。当更多的数据可用时,以及改进推断程序后,矩阵也会跟着更新,例如此后JTT,WAG和LG矩阵的出现。此外,针对特定数据集的矩阵也被引入了,例如线粒体编码蛋白质的mtREV,叶绿体编码蛋白质的cpREV,以及不同二级结构或表面可达性的矩阵。这些矩阵被认为是经验的 (empirical),因为它们是基于许多数据集的平均替代率。这与上述基于机制的模型形成了对比。在机制模型中,选择模型参数以反映关于替代模式的某些假设,并对每个数据集进行估计。值得注意的是,这些矩阵可以分解为两个组成部分:
$$
Q = S \cdot \Pi
$$
其中$S$是描述氨基酸可交换性的对称矩阵,对角矩阵$\Pi$表示氨基酸平稳频率。这20个氨基酸频率可以从每个分析数据中 (+F选项) 估计,因此,氨基酸模型通常是基于非常大的序列摘要信息 ($S$部分) 和数据特异的平稳氨基酸频率 ($\Pi$部分)之间的混合。
跨位点速率差异
上述模型都假设在每个序列位点上具有完全相同的随机过程。这显然也是与实际情况不符的。由于进化过程的随机性质,给定一个系统发育树和一个特定的模型,一些序列位点比其他的位点可能会经历更多的替代。每个位点替代数量的差异遵循泊松分布 (Yang, 1996)。也就说,可以确定,在真实序列数据中每个位点上的替代数分布与模型预期的有本质上的不同,即,与所有位点在同一随机模型下演化的的假设不符。例如,对于一个给定的数据集,与模拟的数据假设一个跨位点的恒定模型相比,经常有大量的不变位点 (即,它们是完全恒定的,沿着系统发育没有经历任何替代)。分枝长度反映了所有位点上的平均替代数。因此,如果整个树中每个位点的平均替代数很高,那么没有经历替代的位点预计将非常罕见。然而,例如在蛋白质序列中,不变位点比预期数多得多。这些位点通常是那些对维持蛋白质的功能或结构至关重要的位点,因此纯化选择可以去除出现在这些位点上的大部分突变。旨在捕获不同位点之间观察到的替代率变异性的模型被称为位点间替代率变化 (ASRV) 模型。为了了解它们是如何工作的,我们首先介绍位点特异的进化速率 (site-specific evolutionary rate) 的概念。考虑一个沿着长度为$t$的分枝进化的位点,比如在JC模型下。由于分枝长度表示每个位点的平均替代数量,因此与沿长度为$t$的分枝演化的位点相比,沿着长度为$2t$的分枝演化的位点预计将积累前者替代数量的一倍。考虑这种情况,在特定位点,祖先为$A$,后代为$C$。当给定分枝长度为$2t$时,数据的似然值为$P_{A,C}(2t)$。这种情况和“沿着长度$t$的分枝进化,而该位点上存在一个位点特异的速率$2$”是等价的。一般的情形,序列$A_1,A_2,...,A_N$ 沿着长度为$t$的分枝,且具有位点特异速率$r_1,r_2,...,r_N$,演化成为序列$B_1,B_2,...,B_N$的似然可以表示为:
$$
\prod_{i=1}^N P_{A_iB_i}(r_i \cdot! t)
$$
通常位点特异的速率是未知的。一种思路是为每个位点赋予不同的速率参数。但是,这将不可避免的增加需要从数据中估计的参数数量,进而会导致非常劣质的统计推断。所以我们通常这样做:将这些不同位点的速率值视为从一个有限速率集合$r_1,r_2,...,r_k$中按概率$\omega_1,\omega_2,...,\omega_k$抽得的数据。也就是说这些速率值具有一个离散的统计分布。然后,我们通过平均所有可能的速率来近似地计算似然:
$$
\prod_{i=1}^N \sum_{j=1}^k P_{A_iB_i}(r_j \cdot! t)\cdot! \omega_j
$$
这样,问题就变成了如何选择这些速率及其概率。Tamura and Nei (1993) 首先提出,对于成对序列,速率是满足伽马分布的。Yang (1993) 提出了假设速率是从一个连续的伽马分布中抽取的,然后计算树似然值的方法。后来为了加速计算,Yang (1994) 假设速率是从离散的伽马分布中抽取的。到目前为止,离散伽马分布是使用最广泛的ASRV分布。参数为$\alpha$和$\beta$的伽马分布平均为$\alpha / \beta$,方差为$\alpha / \beta^2$。通常,使用单位$1$伽玛分布,使$\alpha=\beta$和所有位点的平均速率为$1$。因此,包含这种概率的模型包括一个附加参数$\alpha$,通常用+G表示。在这些模型的离散版本中,通常选择代表性速率,以便每个速率具有相等的概率$1/k$ (Yang, 1994)。此后,虽然出现了基于拉盖尔求积法 (Laguerre quadrature) 来离散化伽马分布的方法 (Felsenstein, 2001; Mayrose et al., 2005),但这些方法很少使用。另外,还有几种伽马分布的替代方案。首先,考虑一个针对不变位点的附加项,生成了+G+I模型 (Gu et al.,1995)。其次, Kosakovsky Pond and Frost (2005) 建议根据使用离散贝塔分布估计的分位数,将伽马分布离散为不同类别。第三, Mayrose et al., (2005) 认为,与使用单一的伽马分布相比,几种离散的伽马分布的混合可以更好地捕获ASRV。还有,Yang (1995) 提出了一个自由参数分布,其中速率和概率都是用最大似然来估计的参数。虽然这种分布非常灵活,可以很好地近似数据特异的特征,但参数也非常多。将ASRV引入进化的概率模型可以提高树推断和分枝长度的准确性,以及许多依赖于进化模型的下游应用 (Yang, 1996) 和量化位点特异的保守分数 (Mayrose et al., 2004)。
混合模型
上述模型假设一个单$Q$矩阵代表了跨越所有位点的进化动力学。然而,选择压力和突变过程可能在位点之间有所不同,从这个意义上说,上述模型又过于简化了进化过程。通过假设有几个可能的瞬时转移速率矩阵$Q_1, Q2, ...,Q_N$,每个位点都与这些矩阵之一相关联。如果我们不知道与每个站点相关的矩阵,我们通过对所有可能矩阵的加权平均来计算似然: $$ P(D|M) = \sum_{j=1}^k P(D|M_j) P(M_j) $$ 其中,$M_j$是通过使用矩阵$Q_j$来定义的模型。这与包含位点特异速率的似然计算非常相似,如上所述,只是在这里我们对各种$Q$矩阵进行平均。这两种情况都被认为是混合模型 (Zhang and Huang, 2015)。混合模型被广泛用于描述密码子的演化。而在等式41中描述的MG密码子模型中,假设所有位点都有一个$\omega$参数,很明显,这个选择系数的类型和强度在不同的位点上是不同的,一些位点经历净化选择 ($\omega < 1$),而一些位点中性进化 ($\omega=1$),或可能处在正选择之下 ($\omega > 1$)。混合模型可以实现对选择强度中的这种变异性直接建模。假设有一组可能的$\omega$值,从而得到一组$Q$矩阵。然后使用上述混合模型的公式计算的似然。针对密码子的混合模型有多种,其中$\omega$值依据所采用的不同分布 (如自由、伽马或贝塔分布) 而变化 (Yang et al.,2000)。使用这些模型,可以通过比较只允许$\omega \leq 1$的$Q$矩阵的零混合模型,和包含$\omega > 1$的$Q$矩阵的备择混合模型的似然,来检验正选择的存在。这些模型也经常被用于推断每个位点的$\omega$的后验估计,从而揭示了在特定选择方式下进化的位点 (Cannarozzi and Schneider, 2012)。
基因家族模型
上述模型由核苷酸、氨基酸和密码子序列的连续时间马尔可夫链构成。然而,马尔可夫模型也可用来描述基因家族的进化动力学。这种模型的最简单形式是分析种系模式 (phyletic patterns),所涉及的数据集是表示 (在一组基因组中) 基因家族有无的矩阵,其中每一行代表一个基因组,每一列代表一个基因家族,如果基因家族$j$的成员存在于基因组$i$中,矩阵中的$i,j$项是1,否则为$0$。这样一个模型的状态是二进制的 ${0, 1}$,因此,可以使用一个$2 \times 2$的$Q$矩阵来建模这些状态的演化。在这样的矩阵中,$λ=Q_{0,1}$是增益率 (gain rate),$μ=Q_{1,0}$是丢失率 (loss rate): $$ Q = \left[ \begin{matrix} - \lambda & \lambda \ \mu & - \mu \end{matrix} \right] $$ 我们注意到,增益事件要么反映了基因家族的从头出现 (de novo emergence),要么是通过水平基因转移的基因拷贝增加。我们还注意到,“1”到“0”的转变反映了一个给定基因家族的所有拷贝的丢失。利用这个速率矩阵,可以显式地推导$P_{ij}(t)$的公式。对这样一个矩阵上施加可逆性,即$\pi_0 \lambda = \pi_1 \mu$导致条件$\mu = \lambda \pi_0/\pi_1$,我们得到了另一种表示: $$ Q = \left[ \begin{matrix} - \lambda & \lambda \ \lambda \pi_0/\pi_1 & - \lambda \pi_0/\pi_1 \end{matrix} \right] = \frac{\lambda}{\pi_1} \left[ \begin{matrix} - \pi_1 & \pi_1 \ \pi_0 & - \pi_0 \end{matrix} \right] = r_Q \left[ \begin{matrix} - \pi_1 & \pi_1 \ \pi_0 & - \pi_0 \end{matrix} \right] $$ 在这里,$r_Q$是一个比例参数 (scaling parameter)。修改其值只会改变沿系统发育树的转变总数,而不会改变增益和丢失的相对数量。如果施加分枝长度反映沿树的平均替代数的约束,应该令$\sum_i \pi_i Q_{i,i} = -1$,进而得出$r_Q = 1/(2\pi_0 \pi_1)$。又因为$\pi_0 + \pi_1 = 1$,所以这个模型就只有一个自由参数:$\pi_0$。上述基础模型在各种研究中被使用和扩展。Hao and Goling (2006) 假设增益率和丢失率是相等的,从而确保了基因组的大小在进化过程中不会有太大的变化。Cohen et al., (2008) 扩展了这个模型,允许不相等的增益率和丢失率,以及使用离散伽马分布引入基因家族之间的速率变化。在这样的模型中,速率不同,但假设所有基因家族中的得失率之比是相同的。随后,通过引入混合模型,减轻了固定增益率和丢失率之比的假设 (Cohen and Pupko, 2010; Spencer and Sangaralingam, 2009)。这类模型的一个有趣的方面是,在分析实际数据时,需要纠正不可观测的数据。考虑一个祖先基因家族全部丢失的例子。在种系模式中,这对应于一个充满$0$的列,反映了一个在现在所有的基因组中都没有的基因家族。然而,在实际数据中,零列从未被观察到,因为种系模式是通过在现存基因组中的同源性搜索来构建的。Felsenstein (1992) 在分析限制性位点存在-丢失数据时已经指出并提出了这个问题的解决方案。具体地说,对一个特定的基因家族 (D) 的可观察数据的概率实际上是以“不为零列”为条件的。设$C_0$和$C_+$分别为“零列”事件和“非零列”事件。我们可以得到: $$ P(D|C_+) = \frac{P(D \text{\,and\,} C_+)}{P(C_+)} = \frac{P(D)}{1 - P(C_0)} $$ 因此,想要的概率$P(D)$是条件概率$P(D|C_+)$和不完全由零组成的列的概率 $1-P(C_0)$ 的乘积。后者可以通过计算零列的互补事件概率来很容易的估计。在上述连续时间马尔可夫模型中,状态“1”表示一个或多个基因拷贝。因此,有可能扩展这些模型来明确地解释每个基因家族中的拷贝数量。理论上,状态的数量是无限的,但在实践中,一个预定义的上界$M$被用来将状态空间转移到一个有限状态马尔可夫链,这样最后一个状态包括所有等于或高于该数的值。在这种情况下,数据被编码在字母表$0,1,2,...,M$。本例中的速率矩阵是新生-死亡模型 (birth-death model) 的一个变体 (Ross, 1996),其中新生率$\lambda$和死亡率$\delta$是单个基因得失的速率: $$ Q_{I,J} = \begin{cases} \lambda & j = i+1 \ \delta & j = i-1 \ 0 & \text{otherwise} \end{cases} $$ 此外,还有一些扩展:(i) 根据新生-死亡-创新模型规定的基因家族的从头出现 (Csurös, 2010; Karev et al., 2003; Librado et al., 2012; Spencer et al., 2006),(ii) 全基因组复制的概率 (Rabier et al., 2014),(iii) 得失率对基因家族数量的依赖性 (Spencer et al., 2006) 和 (iv) 基因组序列覆盖率和注释质量差异 (Han et al., 2013)。
插入缺失模型
虽然替代的概率模型现在常用于重建系统发育树或寻找正选择,但在推断多重序列比对时,通常采用临时的方法 (ad-hoc methods)。这种差异源于建模插入和缺失 (insertion and deletion, indel) 事件比建模替代更具有挑战性,主要是因为将插入缺失事件纳入似然函数中,将导致不同位点独立演化假设的失效。在一篇突破性的论文中,Thorne et al., (1991) 开发了第一个包含插入缺失事件的概率模型 (TKF91)。与替代模型不同 ($Q$矩阵中的状态数为4、20或61),TKF91中这些序列需要作为整体来考虑,不能被视为“独立列”。因此,状态的数量以序列的长度呈指数级增长。大量可能的状态使得显式地直接指数化$Q$矩阵是不可能的。为了降低模型的复杂性,TKF91模型假设,如果序列$i$和$j$之间的长度差大于1,则为$Q_{i,j}=0$。这一假设意味着,较长的长度差异是几个长度为1的插入缺失事件的结果。虽然从我们对插入缺失突变的了解来看,这一假设显然是不现实的,但它是为了让使用这个模型的计算可行,而不得不做出的妥协。在TKF91中引入贝叶斯集成技术 (Bayesian integration techniques) 以及先进的动态规划算法进一步增强了这种概率模型的计算能力。TKF91由同一组研究人员扩展以允许更长的模型 (TKF92, Thorne et al., 1992)。然而,为了克服计算上的挑战,假设重叠的插入缺失永远不会通过进化中发生,这在生物学上也是不现实的。Miklos et al., (2004) 开发了一个完整的长插入缺失模型。虽然这个模型允许任何大小和重叠的插入缺失,但它的计算非常密集型,不能应用于多个序列。最近,Levy Karin et al. (2018) 为这个长插入缺失模型引入了显著的加速方法,并证明使用这样的长插入缺失模型比广泛使用的比对程序能产生更精确的两两比对。统计比对算法旨在使用概率模型对树和比对进行联合推断 (Steel and Hein, 2001)。目前的统计比对工具,如BaliPhy (Redelings and Suchard, 2005) 依赖于隐马尔可夫模型 (HMMs),这也是基于概率的插入确实模型。然而,在基于HMM的模型中,每个树分枝都有一个不同的马尔可夫模型。相比之下,上面提出的插入缺失模型描述了涵盖整个树的单一过程,一组模型参数由所有树分枝共享。虽然基于插入缺失的模型目前落后于替代模型,但它们的发展有望极大地改变各种分子进化的应用,如序列比对算法、各种基因和谱系的插入缺失进化动力学表征 (Chen et al., 2009; Levy Karin et al., 2017a; Lunter, 2007),序列模拟算法 (Cartwright, 2005; Fletcher and Yang, 2009),以及树推断和分子年代测定等下游分析。
更为复杂的模型
随着序列数据和计算资源的增加,更复杂的序列进化推断程序的发展本身就经历了加速的演化。模型开发中有影响力的方向包括:
- 解释树枝之间过程变化的模型。例如,包括只允许在树枝的一个子集上进行正选择的密码子模型 (Yang et al., 2002),和允许进化速率沿树发生变化的模型,例如covarion模型 (Galtier, 2001)。
- 更复杂的混合模型,允许对一组经验氨基酸矩阵 (Quang et al., 2008) 进行平均,或使用贝叶斯方法抽样氨基酸矩阵 (Lartillot and Philippe, 2004)。
- 将蛋白质结构信息与序列进化相结合的模型 (Choi et al., 2007)。
- 将特征信息与序列进化相结合的模型 (Lartillot and Poujol, 2011; Levy Karin et al., 2017)。
- 替代率持续演变的模型 (Lartillot and Poujol, 2011)。
- 允许数据集的不同分区在不同的参数下演化的模型 (Nylander et al., 2004; Lanfear et al., 2016)。
公式编号请参考原文。
全文翻译自:Tal Pupko, Itay Mayrose. A gentle Introduction to Probabilistic Evolutionary Models. Scornavacca, Celine; Delsuc, Frédéric; Galtier, Nicolas. Phylogenetics in the Genomic Era, No commercial publisher | Authors open access book, pp.1.1:1–1.1:21, 2020.