
系统发育冲突的来源
引言
错综复杂的生命进化过程是我们所观察到的遗传序列多样性背后的根本原因。然而,从遗传序列中发现这些进化事件,及其发生的顺序是极具挑战的。本文将描述一些进化过程,如在繁殖过程中从母代到子代的基因传递、基因复制和丢失,和水平基因转移,并证实它们会影响系统发育推断。事实上,如果这些过程被忽视,可能会导致具有较强统计支持的错误推论。
在了解哪些过程会导致系统发育冲突之前,让我们重温同源 (homology) 这一基本概念 (Greek; Homos,相同; logos, 关系) 。遗传序列被认为是同源的 (homologous),如果它们分别有共同的祖先;或被认为仅仅是相似的 (analogous) ,如果没有共同的祖先。同源的定义看起来很简单,但有几层隐含的复杂性。比如理论上有可能所有序列都源自同一祖先。关于两个序列是否同源,涉及到统计检验问题,其中零假设为所观察的相似性只是偶然 (随机) 产生的。
同时,确定同源性并不是比较单个DNA碱基或氨基酸,而是整条分子序列。更准确的说,我们需要关注的是一个基因——抽象的系统发育单元。储存在计算机里的基因是一组字符,其中的字符沿着DNA序列相邻 (也可不相邻,如外显子),不可分割地从母代传递到子代,或者更抽象地说,从供体传递到受体。基因重组 (recombination) 在子代产生过程中对遗传序列进行了重新洗牌 (reshuffling),使不同的基因组片段产生分离,进而使它们的进化历史部分地相互独立。通常我们假设重组只在基因之间起作用,因为基因内部的重组很可能会破坏功能,且多是有害的,会很快从群体清除。
序列同源性通常用两序列之间的距离来衡量,而成对距离又由序列搜索算法 (如BLAST) 来确定。当处理很多序列时,所得到的距离矩阵可以解释为一个完全连通的图 (graph)。顶点是序列,边的权重对应于BLAST分数。经典操作还会为边的权重施以阈值,来生成一个没有权重的、更稀疏的图。然后,使用图聚类算法 (graph clustering algorithm) 和序列重叠 (sequence overlap) 等标准来识别紧密连接的子图 (subgraph),子图所对应的即为同源簇。诚然,区分序列来自同源簇的阈值取决于用户定义的参数,因此具有一定程度的主观性。
同源搜索的结果是将基因根据同源性分成不同的集合,这些集合称为基因家族 (gene family)。基因被分成不同的家族,要么是因为这些基因不够相似,或者因为缺失了某些中间的同源序列样本。毕竟,所有基因有可能都源于同一个基因家族。基因家族是重建祖先的信息宝库,但它们总是带有一些谜团。当决定了两个基因属于同一基因家族,就可以追溯它们的历史,直到它们完成溯祖 (coalescence)。在溯祖完成点 (或称合并点),可能发生过不同的进化事件。首先,最常见的是,相应事件可能与繁殖过程中的细胞分裂有关。在这种情况下,祖先和两个子代都属于同一物种,这两个子代基因位于相同的基因组位置 [称之为基因座位 (gene locus)]。如果此过程伴随物种形成 (speciation),这两个子代基因可能最终出现于两个不同的物种中。我们称与溯祖或物种形成相关的基因为直系同源基因 (orthologs)。其次,溯祖合并点可能对应于某种形式的基因复制 (gene duplications)。这两个子代基因在同一个体基因组的不同座位上共存。这种与复制相关的基因称为旁系同源基因 (paralogs)。全基因组复制是一种特殊的基因复制形式,与之相关的基因被称为ohnologs (为纪念Susumu Ohno)。再者,溯祖合并点还可能对应于某种形式的水平基因转移。祖先和子代基因之一来自同一物种,被称为供体 (donor);另一个子代基因则出现在不同物种 [又称受体 (recipient)] 的基因组中。我们称之为xenologs。最后,由种间杂交产生的基因被称为homeologs。
溯祖和物种分化,是进化的两种不同表达,按时间顺序由远及近的表达进化,即为物种分化;按时间顺序由近及远的表达进化,即溯祖。物种分化是真实的过程,产生生物多样性的基础;而溯祖过程只是理论上的概念,沿着溯祖过程生命体系将逐渐返回单一状态。
一个基因家族中所有基因的进化历史或系统发育关系可以用基因树来描述。我们使用术语——树,来指一个类似树的对象,包括拓扑和分枝长度。与基因树相似,物种的进化关系用物种树来表达。越来越复杂的序列进化模型允许精确地重建基因和物种树。基因重组事件打破了基因家族内不同基因的历史,以及基因家族之间的历史。经过几代之后,谱系关系被抹去,基因彼此独立进化。由于重组,旁系同源、ohnologs、xenologs和homeologs的存在,可能会导致基因树和物种树之间的分歧。因此,系统发育分析方法通常假设基因之间不受重组影响。基因树与物种树的分歧或基因树内对物种树的分歧,即为系统发育冲突 (phylogenetic conflicts)。然而,我们通常对同源性的类型没有先验知识,所以不知道两个基因是orthologs、paralogs、ohnologs、xenologs又或者是homeologs。
此外,一个基因家族中的基因有不同的、独立的溯祖时间。因此,在先前的物种形成事件之前没有完成溯祖的直系同源基因也会导致系统发育冲突。此外,基因之间的自由重组也意味着基因家族有独立的历史,并非沿着同一物种树进化。因此,系统发育冲突也将表现为基因树本身之间的分歧。事实上,系统发育冲突也主要表现在基因树之间,因为物种树是未知的,且不能直接重建。
造成系统发育冲突的过程
遗传序列的变异是由一系列突变完成的,变异可以在群体中传播开来,要么是随机的,要么是因为变异提供了选择优势。影响多个DNA碱基的突变被称为结构性突变 (structural mutations),从几个碱基到数十亿个碱基不等。从基因组中去除部分或添加部分基因的结构性突变分别被称为缺失 (deletions) 和插入 (insertions)。许多生物学过程可以导致在个体基因组内产生新的基因结构。其中一些过程创造了近乎完美的基因拷贝,另一些过程可能只复制部分基因,或结合/扩展不同的基因 (例如外显子洗牌)。相关过程包括逆转录,即一个基因通过逆转录反转,随后插入个体基因组,将移动元素插入基因,或基因的融合和分裂。
尽管创造新基因结构涉及到复杂的生物学过程,但它们在数学可处理模型中的量化需要显著的简化。大多数系统发育模型只考虑基因的完美复制,要么在同一个体中 (基因复制),要么复制/剪切并粘贴到来自不同物种的共存个体中 (水平基因转移)。根据当前的系统发育模型,我们区分:基因起源 Gene origination,涉及新的基因家族的形成;基因新生与死亡 (Gene birth and death),指从基因家族中添加和删除一个基因。此外,种间杂交和不完全谱系分选也是系统发育学需要重点关注的。
基因起源
在三域生物中,有机体的基因组内有多达30%的基因没有已知的同源物,并且可能是最近 (从无到有) 起源的。这些所谓的孤儿基因 (orphan genes) 可以通过创造新的遗传材料来产生,例如由于重组过程的错误、外源基因的获得、或转座机制而产生的基因复制。新遗传材料的产生紧跟着一个快速自适应进化的阶段,导致分歧超过了同源搜索的阈值。
基因家族还可能起源于从头开始的进化 (de novo evolution)。来自非编码区域的随机序列,可能形成神秘的功能位点,并随后受到调控控制。在这种情况下,新基因的产生可能很简单,比如在一个碱基上发生突变来激活下游DNA片段的转录。激活的序列可能幸运地编码蛋白质,并能增强适应性。最初,从头开始的进化被认为是极不可能的,但已经有令人信服的证据证明了它的存在。例如,已经检测到一些人类特异性基因,它们对应于其他灵长类动物的非转录区域。问题是:这些基因究竟是在人类中被打开了?还是在所有其他灵长类动物中被关闭了?
基因新生与死亡
基因家族中基因拷贝数的变化是一个高频率的突变事件。然而,确切的生物学机制很难被发现,因为它们在基因组上位置往往不同。此外,确定基因新生率 (gene birth rates),即单位时间发生造成基因拷贝数增加事件的数量,是很困难的。同时,基因新生和死亡的机制是不同的,所以它们可能会有不同的速率。在概念上,基因新生和基因死亡分别表示基因拷贝数的增加和减少。
基因复制
基因复制 (gene duplication),即个体基因组中基因遗传复本的出现,是生物演化复杂性的组成部分。基因复制在生命树中非常普遍,并且极大地塑造了现存生物的遗传物质。事实上,基因复制是真核生物中最常见的新基因来源。基因拷贝的存在可能会产生有害的,亦或者有益的影响。它存在于一些但并非所有的自然种群个体中,导致一种被称为拷贝数变异 (copy number variation) 的情况。虽然我们对功能和基因组水平上的基因复制有基本的知识,但关于它们的出现和维持的认知是不完整的。导致基因复制的生物学机制有:
-
不等交换 (unequal crossing over) 是指重组过程中的交换发生在染色体上的不相同的位置,然后在减数分裂过程中排列不一致。
-
在复制滑移 (replication slippage) 过程中,DNA聚合酶错误地改变位置,复制部分染色体两次。
-
逆转录转座 (retrotransposition) 是信使RNA逆转录为DNA回到基因组的过程。
-
蛋白质结构域的重复是在许多基因中观察到的一种模式。重复的结构域可能是由外显子洗牌 (exon-shuffling) 引起的,这是外显子在非同源序列之间重组的结果。
-
活性移动元件 (active mobile elements) 的转移也可能导致部分含有重复遗传物质的新基因。
-
多倍体生物的细胞核包含两组以上的染色体。全基因组复制是一种极端类型的突变,在减数分裂期间产生的配子携带整个二倍体基因组,而不是单倍体基因组。全基因组复制会导致所谓的四倍体,这时每条染色体在细胞核中出现四次。全基因组重复再重复将导致八倍体,尽管在连续的全基因组复制之间可能丢失染色体副本,导致染色体拷贝数量不同。全基因组复制在植物中比动物更常见。
-
以类似的方式,在两个物种的杂交期间,两个基因组的融合将导致异源多倍体。
在原核生物中,基因复制的过程还不太清楚,但现在已经有了几种对复制率进行评估的方法。大多数新生的基因将在几个世代内被遗传漂变 (genetic drift) 清除。只有一小部分的新基因的频率会增加,并最终在一个群体中固定下来。在基因复制的情况下,两个基因拷贝可能是冗余的,因此自然选择的压力会松弛下来。进而某个基因拷贝可能再次因缺失突变或功能丧失突变的积累而丢失。更有趣的是,一个基因拷贝可能进化出一种新功能,这个过程称为新功能化 (neofunctionalization)。这两个基因拷贝也可以子功能化,并执行单独的功能,共同完成所有原始功能,或提供更多功能。
全基因组复制是系统发育中的产生冲突的重要因素。例如,在所有颚状脊椎动物的最后一个共同祖先中,两轮全基因组复制 (2R) 使脊椎动物基因增加四倍。2R假说曾经一直存有激烈的争论,但现在已被广泛接受。更重要的是,第三轮全基因组复制已经在鱼类中发生,但证据并不总是结论性的。全基因组复制在植物中更为常见。例如,有证据表明,被子植物共同祖先的基因组被复制了三次。事实上,这可能是被子植物的形态和生态多样性的原因。多倍体增加了生物的复杂性和受自然选择影响的遗传物质的数量。由此产生的表型变异,主要是由于二倍体和多倍体个体之间表达水平的整体差异,可能导致多倍体的选择优势。
由于同义距离的饱和,很难推断古老的全基因组复制事件。最近,利用联合似然估计 (amalgamated likelihood estimation, ALE) 来推断全基因组复制的概率方法已经被开发出来。ALE是基于进化分枝的条件概率的,这大致对应于观察到的进化分枝的频率分布。这些概率可以从基因树的集合中计算出来,或者从自展分析或MCMC分析中产生的树中计算出来。重要的是,基因树的不确定性被考虑在内,也可以评估未观察到的基因树拓扑。
横向基因转移
与基因垂直遗传 (从母代到子代的传递) 相比,生物体也可以通过被称为水平基因转移 (horizontal gene transfer, HGT) 的过程,吸收外来基因,或来自远亲外来基因的变异拷贝。一个水平基因转移事件需要供体成功地释放遗传物质,运输到受体,获得并纳入受体的基因组,最终以一种对受体有益的方式表达。有几个过程能够完成所需的遗传事件。
- 转导 (transduction) 是指通过噬菌体等载体输入病毒DNA,甚至可能是感染的结果。
- 通过质粒的接合转移 (conjugation),涉及细胞与细胞的直接接触的水平转移。
- 转化 (transformation) 是吸收细胞外自由DNA。
- 基因转移因子 (gene transfer agents) 是供体基因组中的类噬菌体元件。基因转移因子有时受到供体的调控控制。它们将供体的随机DNA片段打包,并将其运送给受体。基因转移载体的水平转移与转导的不同之处在于,与噬菌体不同,基因转移因子无法转运自我繁殖所需的基因。
了解不同形式的转移很重要,因为不同机制的传递遗传物质的范围不同,进而它们的特征也不同。有趣的是,HGT在确定DNA作为遗传的分子基础中发挥着关键作用。1928年Griffith的著名实验表明,非毒性细菌菌株可以结合灭活的有毒力菌株的遗传物质,随后导致疾病。1944年Avery等鉴定出DNA是一种通过转化来传递遗传信息的物质。当抗生素耐药性出乎意料地在许多不同的肠菌株中迅速传播时,人们普遍认为,HGT不仅可以在实验室中被诱导,而且在细菌基因组的进化中具有普遍的重要意义。
我们很难对HGT进行直接观察,因此,它发生的证据需要从分子序列本身留下的痕迹中收集。当然,我们期望水平转移的基因在供体和受体之间表现出高度相似,该基因将仅限于最初供体和受体的后代。特别是如果供体和受体亲缘关系较远,无关物种受限制亚群之间表现出的过度相似性,应该引起检测HGT方法的注意。
收集HGT证据的早期研究分析了核苷酸组成和密码子偏好模式。序列特征与基因组中的其他基因存在显著不同时,被归类为最近的转移。在针对细菌和古细菌基因组小规模分析中,检测到的转移DNA数量在0%到近17%之间。而真实的情况很可能还高于此,因为无法检测到发生在具有相似序列特征的物种间的转移。事实上,有越来越多的证据表明,HGT发挥了重要的进化作用,并整体塑造了细菌和古菌基因组,以及它们的多样化和物种形成模式。事实上,很大一部分细菌和古菌遗传多样性是通过HGT获得。原核生物之间高水平的HGT可以导致更像网络状,而不是树状的系统发育关系。直到最近,真核生物HGT的重要性仍然是一个有争议的话题。此外,在整个真菌王国中也观察到大量的转移。
最后,没有可识别同源性的转移可以被解释为基因起源。可能的原因是,转移事件后发生快速分歧、供体物种的基因缺失、错误的同源搜索,或不完整的抽样和序列数据不可得。
基因缺失
基因缺失 (gene loss) 是指从一个基因家族中去除现有的基因。一方面,基因缺失可能是一种突然的突变事件,例如减数分裂期间的不等交换,或移动元件的转位。另一方面,基因缺失也可能是一个缓慢的过程。产生截断蛋白或移码的无义突变,以及影响关键氨基酸位置的错义突变,导致一个基因的初始失活。所谓的假基因化(pseudogenization) 之后是适应度效应较小的一系列缺失事件。无功能基因 (可能是失活的结果),或无功能副本被称为假基因 (pseudogenes)。假基因的数量可能会很大,例如人类基因组的假基因几乎与功能基因一样多。
基因缺失极大地影响了基因组的基因含量,对物种分化的贡献不可小觑,仅次于其它过程,如突变。潜在引起表型的多样性表明,基因缺失可能是一种适应性进化的变异。特别是在细菌和古菌中,基因组大小是一个强适应度决定因素 (少即是多假说 less-is-more hypothesis)。此外,高水平基因缺失也会发生在物种内。
杂交
物种偶尔也由从两个不同祖先物种的杂交而来,这一事件被称为物种间杂交 (inter-species hybridization)。种间杂交在植物中尤其常见。由此产生的基因组和表型特征揭示了这两个祖先的来源。表现出物种间杂交的物种历史有重新连接的分枝,因此,它们不是树状的,而是对应于更一般的网络,称为有向无环图 (directed acyclic graphs)。各基因树可以采取不同的拓扑结构,这取决于哪些基因副本被保留或丢失。
小麦是我们最重要的主要作物之一,已经种植了一万多年。小麦的演变包括一些杂交事件,它的基因组由三个密切相关的亚基因组组成,通常被表为 (AABBDD)。野生乌拉尔图小麦 Triticum urartu (AA) 和拟斯卑尔脱山羊草Aegilops speltoides谱系 (BB) 的首次杂交产生了一种异源四倍体,其中杜伦Durum小麦是它的直系后代。随后,与野生粗山羊草Aegilops tauschii (DD)的第二次杂交事件形成了今天的异源六倍体基因组。
此外,对转录组数据的分析显示,小麦亲属中普遍存在古老的杂交事件。检测杂交事件可以使用杂交指数 (hybridization index),它衡量的由杂交事件造成系统发育冲突的可能性,而不是不完全的谱系分选。在酵母谱系中也发现了一种古老的种间杂交。研究者提出,由此产生的杂交种被迫进行随后的全基因组复制,以恢复繁殖能力。另一种形式的杂交是与病毒的重组。同时被两种病毒株感染的宿主细胞可能会组装出混合起源的新病毒颗粒。一些遗传物质可能来自第一个菌株,其他遗传物质可能来自第二个菌株。
不完全谱系分选
下面我们将描述一个与基因复制和缺失、水平基因转移或杂交有根本不同的过程,因为它在群体水平上发挥作用,并导致直系同源基因之间的冲突。等位基因 (allele, allelomorph的简写) 是一个特定基因座位上基因序列的变体。这里我们指的是同一物种的不同直系同源基因,但在不同的背景下,等位基因也可以是不同的核苷酸、氨基酸,甚至整个染色体。
不同的等位基因可以在一个种群中共存很长一段时间,跨越物种形成事件。从概念上讲,二叉分歧的物种形成过程将位于特定座位的等位基因分选到第一个或第二个子物种。存在较大变异等位基因之间,由于它们可能部分地参与了物种分化,将会被完全分选,进而在子物种中不再存在不同的等位基因。相比之下,由于重组,共存的等位基因可以在物种形成事件中不完全地分选,因此它们会共存于两个子物种。例如下图1所示的,在第一子物种中,与包含蓝色等位基因的个体相比,包含红色等位基因的个体与第二子物种更相近。当然,由于重组,误导性亲和关系只在这个特定的座位观察到。当分析更多的座位时,一个物种中的个体之间会比与其它物种的个体关系更密切。
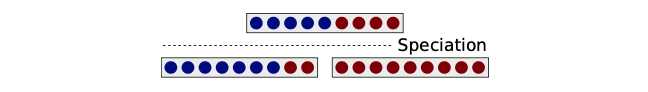
分选 sorting,即分类、挑选、整理。这里特指物种分化过程中,等位基因在不同子物种中的分配。
上述过程被称为不完全的谱系分选 (incomplete lineage sorting, ILS)。在多个连续的物种形成事件中共存的不完全分选的等位基因可能会导致系统发育冲突,因为相应的基因树支持一个与物种树不同的拓扑结构。决定ILS的流行程度的因素是什么?首先,重组是一个先决条件。此外,在一个特定的座位,当多个等位基因在一个种群中共存,且多于一个的等位基因具有较高频率,那么不完全分选就容易发生。一个众所周知的测量方法是基因座位的杂合性 (heterozygosity, $H$),它表示的是当从群体中随机抽样时,取到两个不同等位基因的概率。因此杂合性越高,ILS越可能发生。
对于具有中性变异的单倍体种群,杂合性$H$在每一世代中因遗传漂变而降低, $$ \Delta!H_{D} \approx -\frac{1}{N}H $$ 其中$N$是种群的大小,$\Delta!H_{D}$表示由漂变带来的杂合性变化,且为负值,所以群体大小较大时,杂合性降低的就越小,进而保持高杂合性,那么ILS就普遍。
众所周知,随机遗传漂变主要在较小的群体中对等位基因多样性产生影响,即等位基因在子代中被随机的清除。
杂合性$H$又在每一世代每座位中突变速率$u$增加而降低, $$ \Delta!H_M \approx +2u(1-H) $$ 所以,ILS在一个高突变率的大群体中将会比较普遍。只有这样,才能抵消遗传漂变对它的清除作用,并造成足够的变化。此外,等位基因的ILS,需要在多个物种形成事件上共存,才能导致基因树与物种树之间的分歧。在这种情况下,仅仅是种群规模大是不够的,还需要物种形成事件之间的间隔世代数够低,才会出现不完全分选。也就是说,当物种树内部分枝较短时,不完整谱系分选的发生率更高。
ILS来源于谱系 (lineage) 这个术语,表示来自一个共同祖先。当沿着时间轴向后 (从现在向过去) 看这个过程时,谱系的概念特别重要。这个时候,ILS表现为:同一个物种中的不同等位基因谱系,并不在该物种中完成溯祖 (coalesce),而在该物种的祖先中完成。因此,深度溯祖 (deep coalescence) 一词经常被用来描述ILS。如果溯祖事件涉及一个谱系最终演变为不同物种,深度溯祖只会导致系统发育冲突。
系统发育描述
开发合适的模型是理解观察结果和收集验证假说的证据的关键。首先,我们提醒自己,只有在基因没有从母代共同传递给子代时,系统发育冲突才是一个问题。例如,线粒体基因组不经重组而传递,这可以简化系统发育分析。然而,对线粒体的分析并不令人满意,因为线粒体只包含有限的基因,出现统计误差是可想而知的。相比之下,虽然真核核基因组包含大量的信息位点,但又受到重组的干扰。
同样,包括细菌和古菌在内的数据集也不能被分析,因为它们也显示了由水平基因转移结合同源重组的重组痕迹。由此产生的非直系同源基因的存在是一个普遍问题。一种分析非纵向传递基因的方法是,串联假定的直系同源基因,并希望相应的系统发育信号超过旁系同源基因和xenologs等带来的虚假的、错误的信号。然而,这个策略忽略了很多数据。充分利用包括重组基因在内的数据,需要采用适合分析旁系同源基因、ohnologs、xenologs和homeologs的方法。
同源群大小
历史上,寻找同源基因只在物种内进行。这里,同源群 (homologous group) 将被用来表示一个物种内的一组同源基因。不使用基因家族 (gene family) 这个词,因为基因家族通常涉及多个物种。早期的方法试图描述同源群大小的频率分布,发现它遵循以长尾为特征的幂律分布 (power law distribution)。特别是,具有10个或更多基因的同源群,比中等大小的同源群的丰度更高。值得注意的是,这种分布在细菌、古菌和真核生物中的分布非常相似,这表明导致产生和去除遗传物质的潜在过程有共同的普遍特征。
利用随机线性生灭过程 (stochastic linear birth and death processes) ,可以提高我们对幂律分布在描述同源群大小频率中的作用的理解。线性生灭过程是一个描述整数状态变量演化的随机过程。这里考虑的是一个数字系统,包含数字:$N_t \in {0,1,2,...}$,表示在时间$t$时单位的数量,并按以下规则演变:
- 一个单位产生的子单位彼此完全独立。
- 在时间$t$处存在的一个单位,该单位在时间间隔$dt$内,以概率$\lambda!dt + o(dt)$进行二元分裂方式的新单位产生,并以概率$\mu!dt + o(dt)$进行消亡。
- 群体中的所有单位都具有相同的新生率 (birth rate $\lambda$) 和相同的死亡率 (death rate $\mu$)。
详细地说,新生事件将在群体中增加一个单位,死亡事件从群体中移除一个单位。等待下一次新生或新生事件的时间独立且相同分布 (指数分布)。预定义的新生率$\lambda$和死亡率$\mu$,在所有单位中共享,与单位数量$N_t$无关。等待下一个事件的时间间隔越小,群体中存在的单位就越多。
在当前的讨论中,生灭过程的单位是同源群内的基因,新生事件对应于基因复制或HGT事件,死亡事件对应于基因缺失事件。如上所述,在生灭过程中瞬间产生相同的基因拷贝,只是基因新生与死亡生物学过程的粗略近似。首先,死亡率通常被估计高于新生率。其次,数学分析表明,我们不能仅用线性生灭过程来解释观察到的幂律分布。也就是说,没有哪一组新生和死亡参数可以很好解释同源群大小频率分布的长尾特征。相反,我们需要放松关于独立的假设,并采用广义的生灭过程,其中的新生率和死亡率可以取决于群体的单位总数量。当使死亡率随同源群的大小而降低时 (大同源群死亡率高,小同源群死亡率低),就能解释观察到的长尾特征。
幂律分布也可以用于描述,考虑新同源群产生 (origination of homologous groups) 的生灭过程的稳定分布。具体地说,该模型是两层随机过程的叠加。首先,一个类似于纯新生过程的随机过程,其死亡率为零,描述了同源群的产生。因此,同源群以给定的速率随机复制。当然,同源基因的数量在不同同源群之间可能会有所不同。因此,这个过程涉及到不相同的单位,而不是一个经典的生灭过程。其次,对于每个同源群,单独有一个具有参数$\lambda$和$\mu$的线性生灭过程描述了各自同源群中的基因数量。这两层过程的平稳分布 (stationary distribution),当$\lambda \leq \mu$时,表现为扩展指数分布 (stretched exponential distribution);当$\lambda$ > \mu$时,则表现为幂律分布。平稳分布的存在,因为当最后一个基因死亡时,同源群会被去除。总之,当我们独立地处理同源群内的基因时,我们无法得到观察到的幂律分布。然而,同源群大小的频率分布可以用相对简单的随机过程来描述,如广义生灭过程 (gernal birth and death processes),或使用同源群的两层随机过程对其起源进行建模。需要注意的,这两种模型都独立地处理不同的同源群。
与此同时,不同于基因家族,同一物种内不同的同源群,它们的进化历史是相关的,因为它们都沿着同一物种树向前演化。此外,属于同一基因家族的同源群,在近源物种间要比在远源物种间具有更相近的大小。在这方面,已经开发出了gain-duplication-loss模型,使用从串联比对推断出的固定物种树,来评估基因家族内同源群大小的可能性。在该模型中,gain是水平基因转移的代表,但由于只考虑基因的计数,而不考虑基因树本身,因此转移基因的源头是不清楚的。我们所能观察到的,只是基因总数的增加。忽略基因树,可能会导致基因转移推断中的偏差。速率的变化可以用一个离散的伽马分布来解释,类似于替代模型中进行的处理。此外,还可以使用分枝方式 (brach-wise) 的gain率、duplication率和loss率。对古菌和细菌的应用表明,在被分析的两个域的新生率和死亡率是相似的,而且死亡率似乎大于新生率。
多序列比对
测序技术的进步,以及在同源搜索中使用的聚类和比对算法的改进,已经导致许多具有可用序列数据的基因家族被识别。对遗传序列本身的分析比仅仅分析其数量要有趣得多。序列数据通常以多序列比对的形式呈现 (图2),其中包含大量信息。一般来说,每个基因家族有一个多序列比对,每个序列都有相应的物种标签和他们自己的标签 (identifier),因为每个物种可以有更多的基因。通常,比对不包含关于基因同源关系和基因座位的信息。我们只使用整数形式的基因座位来编码同源性的类型,而不是相对位置。也就是说,座位1不一定是远离或接近座位2的。然而,同一座位的基因被认为是直系同源的,而不同座位的基因被认为是旁系同源基因、ohnologs,xenologs或homeologs。此外,基因家族中常见的多序列比对只提供来自参考基因组的信息,而无关基因的遗传变异。详细地说,它们不包含来自不同个体的序列 (只有多个个体的信息才能反应遗传变异)。基因组这个术语使用常常带有矛盾性。首先,基因组可以指一个个体中存在的完整的遗传物质。其次,以一种更抽象的方式,基因组可以指一个物种、一组物种、或一组个体的遗传物质;例如,“人类的基因组”或“古细菌基因组”。即使我们知道这个词同音同形异义,我们也不能完全避免它。然而,我们的目标是明确地描述系统发育冲突的来源。这就是为什么我们关注个体本身,例如,我们审视“存在于个体中的基因” (而不是在基因组中)。
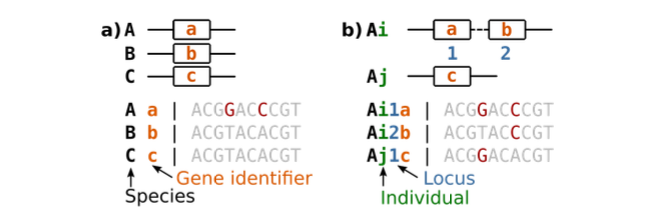
对于一个给定基因家族的多序列比对,我们寻求连贯地描述可能的进化历史。这个任务涉及到一个基因树的构建,最重要的是,树节点所对应的进化事件类型。我们已经简要地讨论了一些可能在基因树节点上发生的事件类型,并用目前的进化方法解释。例如,基因树节点可归类为基因复制或水平基因转移,并添加与基因缺失对应的未观察到的节点,使观察到的基因树与物种树保持一致;这个过程称为基因树/物种树调和 (gene tree-species tree reconciliation)。大多数基因树-物种树调和方法涉及两个步骤:(1) 用最大似然方法重建基因树;(2) 然后用最大简约法最小化基因复制和缺失的总数。
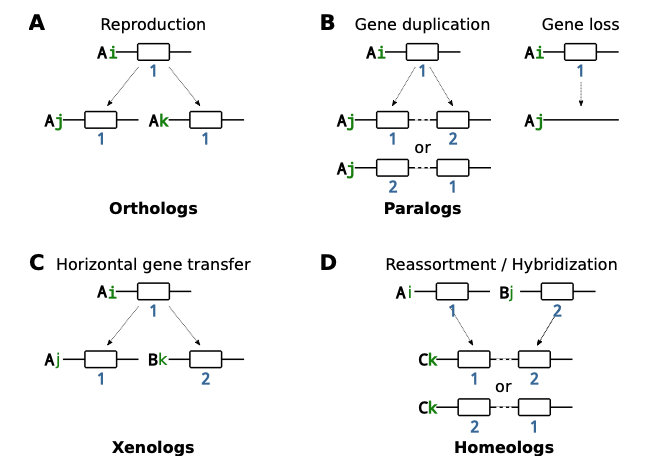
现在我们将详细介绍基因复制和缺失、水平基因转移和杂交,如何影响基因和基因组的细节 (上图3)。繁殖过程中的细胞分裂,或从时间上向后看的溯祖,是指在一个物种中由一个个体产生两个个体,它们都包含原始的基因。这些基因的座位保持不变。而基因复制将同一基因的副本添加到新一个产生的子代个体中。新的基因副本被插入到一个新的座位上。副本基因序列与正本是相同的,所以我们不知道在新的基因座位上发现的是哪个基因。在模型设计过程中,我们可能必须考虑到这个组合学上事实。全基因组复制对应于大规模、联合的基因复制,但可以用简单的基因复制来描述。
HGT将一个基因拷贝插入到一个不同的共存物种的基因组中的新基因座位上。有人认为,水平转移的基因可以接管受体现有基因的功能。在这种情况下,对已存在基因拷贝纯化选择的缺失,会导致已存在基因拷贝的分化或缺失。其结果是一个被称为替代转移 (replacement transfer) 的事件。因为它们的生物学相关性,有一些模型只允许替代转移,也因为替代转移对应于一个特定的拓扑结构改变,称为子树修剪 (subtree pruning) 和重新移植 (regrafting)。
与上面关于全基因组复制的处理类似,种间杂交可以被模拟为发生在两个祖先物种间的、大量联合的水平基因转移事件。全基因组复制和种间杂交事件不仅在基因树上可见,而且在物种树上也可见。最后,基因缺失只是从基因组中删除一个基因。在我们重建的基因树中,不能直接观察到基因损失,因为与缺失事件对应的树枝被从树上修剪掉了。然而,基因缺失是系统发育模型的一个重要组成部分,因为它可以与其他过程一起,或单独造成系统发育冲突。
上述考虑的一个结果是以下想法:如果我们有多序列比对中序列的基因座位信息,我们可以大大减少解释数据的可能进化历史的数量。直系同源基因必须来自不同的基因组的,同一基因座位;基因复制和缺失涉及,来自同一基因组但不同座位的基因;水平基因转移涉及,来自不同物种的 (可能是) 不同座位的基因。注意,扭转这个论点,解释上述事件的概率模型是对于给定多序列比对中基因同源关系。除了阐明基因树之外,探测直系同源基因、旁系同源基因和xenologs具有重要的应用意义。此外,我们只知道一个描述基因座位共线性的模型,即座位的物理协同定位 (physical co-localization of loci)。因此,基因复制和缺失可以影响跨越多个座位的片段。
总结与讨论
总之,基因这个词粗略地表示作为整体传递的一段遗传序列。如果两个基因具有可检测的共同祖先,那么它们彼此同源。一组同源基因被称为一个基因家族。一个基因家族通常跨越许多物种,且每个物种中可以有不止一个该家族的基因。一个基因家族的系统发育历史可以用基因树来描述。基因的谱系是从该基因到基因树的根的路径。两个基因的同源性类型是由这两个基因的谱系在完成溯祖合并时发生的事件来定义的。我们讨论的同源性类型包括:1) 与繁殖过程中细胞分裂有关的直系同源;2) 与基因复制相关的旁系同源;3) 与全基因组复制相关的ohnologs;4) 与水平基因转移相关的xenologs;5) 以及种间杂交相关的homeologs。
系统发育冲突是指基因树与物种树的完全或部分分歧。物种树通常不为所知,因此,系统发育冲突要么表现为单个基因树与物种树的分歧,要么是不同基因树之间的分歧。上述同源关系都可能导致系统发育冲突。例如,繁殖与重组和突变相结合,会导致不完全谱系分选,进而产生误导的拓扑结构。
基因树之间冲突的一个先决条件是重组。相比之下,共同传递的基因不会表现出冲突。因此,基因组结构,如染色体、质粒或核质分隔,是一个重要的因素。人们对基因作为系统发育单位的怀疑正在提升。例如,可以使用更小的单位——外显子。此外,关于重组模式的知识可以帮助区分系统发育重建错误和真正不同的基因树。如前所述,线粒体基因之间相互冲突是出乎意料的。
在系统发育分析中,正确地描述同源关系是势在必行的,然而,实际情况中系统发育冲突的丰富程度是一个有争议的问题。多年来,不完全谱系分选的重要性一直是一个有争议的话题。此外,推断的基因复制和水平基因转移的数量很大程度上依赖于所使用的物种树。虽然这是意料之中的,但在解释涉及系统发育冲突的推论时必须谨慎。一般来说,识别系统误差和统计误差是困难的,如假设同源线粒体基因之间的系统发育冲突可能主要是由统计错误或系统错误引起的。
另一个问题是这些引起系统发育冲突的原因的相对重要性。当连续的物种形成事件之间的世代数较低时,ILS的概率就会很高。如果以世代数测量的平均分枝长度从物种树的根下降到树的末端,ILS在较近时间范围内 (末端附近) 更为普遍。如果我们假设物种树是根据纯新生模型进化的,这个假设不会被满足,因为树上的平均分枝长度与分枝的位置无关。源自线性生灭过程的树分枝长度分布没有解析解。然而,模拟分析的证据表明,当死亡率从零增加到更接近新生率的值时,内部分枝长度相比于比末端分枝长度会增加。这种影响,死亡率越接近新生率,就越明显。Zhaxybayeva和Gogarten 假设生命树是根据溯祖模型进化而来的。溯祖模型假设总种群大小是恒定的,并且到下一次溯祖完成的时间 (随着时间向根移动) 呈指数分布。因此,分枝的平均长度向根方向增加。在这种情况下,以及生灭过程中,ILS的相对重要性从物种树的终端到根部逐渐减少。当然,我们只能假设分枝长度的分布,因为生命之树是未知的。
如果重建方法出现系统误差,也会出现虚假的系统发育冲突。例如,跨位点或跨基因组成的异质性可能会导致拓扑误差。一般来说,序列距离的饱和会导致长分枝吸引的非自然状况。此外,在同源搜索过程中做出的决策也可能会引起虚假的系统发育冲突,并大大影响基因缺失的识别。特别是,如果没有正确地检测到基因起源,那么在邻近物种中缺乏与新基因家族相关的基因,可能会被错误地归因于大量的基因缺失。同样,未检测到的基因拷贝可能被误解为基因缺失。
与这个主题相关的另一个方面是,在所有的系统发育分析中,大多数物种仍然未被采样。因此,有学者分析了来自生灭过程的树,包括树终端不完全采样的概率。本质上,抽样概率对应于假定完全抽样的新生率和死亡率的转换。有趣的是,一个相对简单的计算表明,我们应该期望大多数HGT的供者是灭绝或未采样物种的成员。
最后一个示例显示了在系统发育推理中使用生灭过程的一个很大优势。关于生灭过程的数学研究有着悠久的传统,许多性质已经被解析地推导出来。例如,随时间变化的预期单位数和相应的方差都是已知的。此外,还推导出了新生事件的概率密度和重建树起源时间的分布。这些知识可以通过时间图引人注目地总结在所谓的谱系中,时间图显示了随着时间在生灭过程下进化的树平均谱系数。还有,基因死亡的概率和在给定时间内没有变化的概率,可以用来计算基因树在约束物种树的框架下,按生灭过程进化的概率。然而,仍然很难模拟上述基因树,其条件为每个物种有特定数量的基因,或基于该过程的起源时间。我们可以使用前向过程和拒绝抽样,但对于较大的树,计算时间是巨大的。当考虑到水平基因转移时,这个问题变得更加困难。主要原因是生灭过程的假设不再得到满足。也就是说,由新生事件产生的子单位并没有完全独立地进化。
无论如何,对ILS、基因复制和缺失以及HGT的联合处理都是必须的,但系统发育和种群遗传模型的统一将提高我们对生命树的理解和重建生命树的能力。一般来说,遗传变化跨越了三个阶段:(1) 通过突变在个体基因组中起源,(2) 然后在各自种群中固定下来,(3) 最后得以维持。基因复制的动力学略有不同,因为副本的命运被紧密联系起来,并由在固定阶段或之后积累的变化决定。Innan 和Kondrashov (2010) 认为,为了理解这些过程,我们必须在群体层面上检查基因拷贝的遗传变异。系统发育模型,结合对基因复制和缺失的描述,以及HGT与对种群变异进化的描述,如多物种溯祖模型,可能在这方面发挥重要作用。特别是,因为它们将允许更精确地识别同源性的类型,这是一个重要的信息,可以帮助解决完整的动物物种树。
在这方面,Rasmussen和Kellis (2012) 的三树模型 (three-tree model) 被视为一个相当大的方法进步。此外,推断出杂交事件现在也成为可能。杂交是指已经分化的物种的个体成功地有了后代。杂交种可能是一个新的独立物种的创建者,将物种树转化为系统发育网络。杂交也可以解释为影响大规模水平基因转移。另一方面,我们引入了一个更一致的三树模型,但假设同一单倍型上的同源基因之间没有重组。这两种三树模型都使用了超度量的、类时间的树,但速率修改器可以用来解释不同的分子钟。此外,这三树模型并没有解释物种树上的基因起源。这对于仅限于少数物种的基因家族来说,可能是有问题的。例如,考虑两个来自同一同源基因家族的基因,它们存在于人类和大象中,但在所有其他哺乳动物中都没有。这难道不是仅仅由基因缺失 (或HGT) 引起的系统发育冲突吗?
系统发育树和网络是各种杰出的概率方法的基础。即便如此,它们也可以伴随着受到机器学习社区启发的方法。例如,K-means聚类算法已经应用于由Rasmussen和Kellis的三树模型模拟的数据上,并创建可识别同源区域的分类器。注意,分类器不是一个独立的方法,因为它是在使用所讨论的概率方法模拟的数据上训练出来的。总之,我们可以很自然地期望不同的基因家族讲述不同的关于该物种树的故事。看到对系统发育冲突的三个最重要的原因的处理是如何以一种成功和决定性的方式结合起来的,这将是令人兴奋的。
全文翻译自:Dominik Schrempf, Gergely Szöllösi. The Sources of Phylogenetic Conflicts. Scornavacca, Celine; Delsuc, Frédéric; Galtier, Nicolas. Phylogenetics in the Genomic Era, No commercial publisher | Authors open access book, pp.3.1:1–3.1:23, 2020.